Abstract
Amyotrophic lateral sclerosis (ALS) is a genetically heterogeneous disorder that shows a characteristic dichotomy of familial forms typically displaying Mendelian inheritance patterns, and sporadic ALS showing no or less obvious familial aggregation. While the former is caused by rare, highly penetrant, and pathogenic mutations, risk for sporadic ALS is probably the result of the combined effects of common polymorphisms with minor to moderate effect sizes. Owing to recent advances in high-throughput genotyping and sequencing technologies, genetic research in both fields is evolving at a rapidly increasing pace making it more and more difficult to follow and evaluate the most significant progress in the field. To alleviate this problem, our groups have created dedicated and freely available online databases, ALSoD (http://alsod.iop.kcl.ac.uk/) and ALSGene (http://www.alsgene.org), which provide systematic and in-depth qualitative and quantitative overviews of genetic research in both familial and sporadic ALS. This review briefly introduces the background and main features of both databases and provides an overview of the currently most compelling genetic findings in ALS derived from analyses using these resources.
Introduction
For amyotrophic lateral sclerosis (ALS), as well as for many other neurodegenerative disorders such as Alzheimer's disease (Citation1) and Parkinson's disease (Citation2), familial aggregation was recognized as a salient feature decades before any of the underlying biological and biochemical properties were known (Citation3). In many cases, the identification of specific, disease-segregating gene mutations has first directed the attention of molecular biologists to certain proteins and pathways. The breakthrough in understanding ALS pathogenesis started with the discovery of autosomal-dominant mutations in SOD1 on chromosome 21q22 (encoding soluble copper/zinc superoxide dismutase 1) in 1993 (Citation4). In the last decade, 12 additional genes causing various forms of ALS have been identified (), and it can be anticipated that the number of ALS-causing mutations will increase further with the application of recently developed massively parallel sequencing techniques.
Table I. Genes proposed to cause Mendelian forms of ALS.
Another similarity to other neurodegenerative diseases is the prominent dichotomy of these rare, familial forms of ALS following Mendelian inheritance (5–10% of all cases) alongside seemingly ‘sporadic’ forms of the disease showing no or less obvious familial aggregation, with no clear clinical or pathological distinction between the two. While a small percentage of the sporadic ALS cases have also been found to harbour mutations in the known Mendelian ALS genes, the aetiology for the majority remains largely unknown. It is likely that there is a contribution from common genetic variants (polymorphisms) that are also present in the healthy population (with frequencies > 1%). Each of these polymorphisms probably exerts only a small effect on ALS risk (odds ratios (ORs) in the order of ∼1.2), so that it is via their combined action that they impact ALS susceptibility. Because of the small individual genetic effect sizes, thousands of subjects have to be tested to uncover these risk genes with sufficient confidence (i.e. to achieve genome-wide significance which is commonly defined as p-values falling at or below ∼5 × 10−8). Several hundred candidate-gene association studies have been performed in the field of ALS genetics during the last two decades, yielding mostly inconclusive results. In recent years, genome-wide association studies (GWAS), which can assess several million polymorphisms across the genome in one experiment, have suggested several new ALS risk genes, most of which are still awaiting independent follow-up and replication.
The growing amount of information in the field of ALS genetics is becoming increasingly difficult to follow, evaluate and interpret. This is true for both Mendelian and non-Mendelian, genetically complex ALS. To address this problem, the ALSoD and the ALSGene databases have been developed independently. Both databases aim to provide systematic and in-depth overviews of the respective ALS genetics fields. The aim of this review is to introduce both resources by outlining their complementary goals and methods, as well as by summarizing the most interesting recent findings of each project.
Assessing Mendelian mutations and rare variants in ALS: the ALS Online Genetic Database (ALSoD)
Although the first human disease-causing mutation was identified in haemoglobin in 1949, the pace of discovery was such that the relationship between gene variations and the diseases they caused could be easily remembered. As the numbers grew, it became important to store such information systematically, and so the first genetics database came about in the1970s when McKusick created a catalogue of mutations and inherited diseases, known as “Mendelian Inheritance in Man”. A version of this database was later developed for online access (Citation5), and is now known as OMIM. By 1994 there were regularly updated online lists of gene mutations as well as gene-specific databases. Key to the success of such resources was a universal and logical mutation naming convention based on amino acid changes (Citation6,Citation7).
Mutation databases of human genes are now becoming more prominent and important in all areas of health care. Published and unpublished mutations are typically curated in locus-specific databases (LSDB) (Citation8), and apart from providing vital information on protein function and structure, databases of mutations causing Mendelian disease play a fundamental role in research, diagnostics, and genetic health care (Citation9,Citation10). The existence and regular upkeep of these databases is often vital to a broad range of research fields.
Following the identification of ALS-causing mutations in SOD1 there was a great focus of research on this gene. As the number of mutations increased and the range of phenotypes expanded, the need arose for a database that might reveal new genotype-phenotype relationships, keep track of the different disease-causing genes, and aid the recording of gene variations that might not be published. From this came the ALS Online Database, ALSoD, an open source data repository designed for sharing clinical and genetic data, developed by a consortium within the World Federation of Neurology (Citation11,Citation12). It provides both the scientific community and wider public with up-to-date information on ALS-related genes identified from family studies, candidate gene studies or genome-wide association (). The functional importance of such a database is underscored by the appearance of other similar websites, e.g. an ALS mutation database recently constructed as part of the Life Science Integrated Database Project conducted by the Japan Ministry of Education, Culture, Sports, Science, and Technology (Citation13). This contains their original experimental results as well as published data extracted from scientific journals. This database is expected to play a complementary role to the ALSoD website, especially in collecting genetic variations commonly found in Asia. The ALS Online Database website was initially located at http://www.alsod.org and is now located at http://alsod.iop.kcl.ac.uk, but it remains a World Federation of Neurology resource, not tied to any institution. This change in web address was a simply the result of the need for huge data storage.
Figure 1. ALSoD homepage.

Aims
ALSoD (freely available at http://alsod.iop.kcl.ac.uk) is designed to allow easy access to information about ALS-causing gene variants and their phenotypic effects. Because genotype-phenotype correlations are usually clearest for Mendelian disease genes, and the database has been in existence for several years, much of the focus is on familial ALS, but all currently known ALS-causing genes are represented. Where relevant there are links out to an appropriate database resource. For example, the evidence for association at a given gene is provided by a link out to the ALSGene database where meta-analysis and other explorations are possible (see below), and predictions of SOD1 mutation effects on structure and function are provided by a link to a SOD1 protein structure database at UCL, whereas phenotype information on TARDBP, FUS or OPTN mutations is accessed locally. Mutation hot and cold spots, country-of-origin effects and literature and bioinformatics resources are also included within ALSoD.
Methods
ALSoD allows users to submit new gene, mutation and patient data. It also has various tables in the database schema, storing data on sequence mutation data on genes, and anonymized patient data including phenotypic features and country of origin. Published and unpublished data are continually curated by the database administrator and registered ALS researchers. Published data are obtained by regular PubMed searches, and screening of cross-references from relevant publications. Unpublished data are uploaded by users (as described above) or made available to the ALSoD investigators by collaborations, and are published on the ALSoD website after consistency checks performed by the database administrator. There are currently 12 contributors from various international institutions.
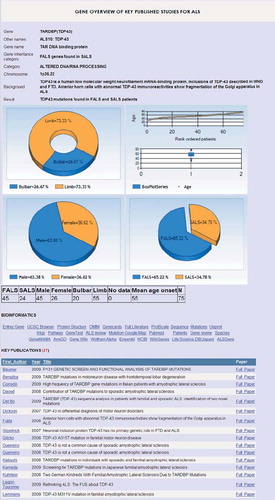
To allow integration of different databases through one gateway, ALSoD uses unique identifiers to systematically link to other bioinformatic tools and comprehensive databases. Selecting a link automatically interrogates the relevant third party website with the appropriate genetic information. Thus, it is possible to identify the most up-to-date list of genes implicated in ALS, explore their interactions and pathways, and review the relevant literature. Customized and routine analyses of mutational relationships with phenotype are possible, either for genes with phenotypic data, such as SOD1, TARDBP, FUS, SETX, and others, or as a general overview of familial and sporadic phenotypic patterns ().
Figure 2. Genotype-phenotype summary for TARDBP (coding for TDP43). Gene summaries are displayed on selection. More detailed customised analyses are also possible allowing specific questions to be asked of the database.
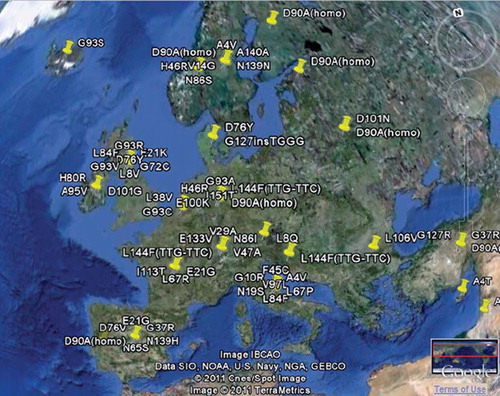
The mapping of gene variants on Google Earth allows researchers or patients to view the geographical distribution of reported gene variations associated with ALS (). The same computational method allows us to map the origin of users and show which countries predominantly access ALSoD.
Figure 3. An example of a geographical overview of the mutation distribution of SOD1. Geographical analysis is available for most FALS genes. More sophisticated map overlays are planned.
For users with their own association data, an on-the-fly analysis is available to combine the data available in ALSoD with unpublished user data that can be confidentially uploaded. The user data is formatted accordingly before upload and the result is fed back in minutes without storing users’ data on the database.
Results
Currently, ALSoD displays information on 73 ALS genes (17 for familial ALS), 298 mutations and 419 patients (343 with age of onset data and described in analyses below). Typically there are more than 100 unique users visiting ALSoD each day, and in November 2010 the pages were accessed more than 18,000 times. Data can be displayed or interrogated online or downloaded into a spreadsheet. For genes, the mutation type, sequence change, original and mutated amino acid, functional annotation and exon number are reported, with a link to the predicted mutant structure. For patients, the gene, mutation and mutation type are reported along with the patient's sex, country of residence, ethnicity, age at onset, clinical pattern, survival data and family history.
Analysis of the record of Mendelian mutations in which one would expect a 50:50 male: female ratio, shows that the actual recorded sex ratio is 172:119, which is almost exactly the 3:2 ratio commonly reported for ALS in general from clinic-based studies. While for 250 patients there is a family history of ALS, 84 patients have none despite a Mendelian mutation, and nine record it as unknown. Those without a family history are not skewing the sex ratio, however, as for those with a family history the sex ratio is 120:88, which is still very close to 3:2.
The mean age of onset overall is 45.7 years. Excluding those with juvenile onset (for example with ALS2 gene mutation), mean age of onset is 46.2 years. The mean age of onset for those with a family history is 42.8 years while for those without it is 53.3 years, which is very close to the mean age of onset recorded in clinic based studies, and suggests that a family history is associated with a younger age of onset. However, for those with SOD1 mutation, mean age of onset is 47.2 years (n = 125), which breaks down into 47.9 years for those with a family history (n = 105) and, surprisingly, 41.7 for those without (n = 19). For TARDBP mutations (codes for TDP43 protein), mean age of onset is 55.5 years (n = 75) and for FUS mutations, 50.4 years (n = 5). There are 31 different TARDBP mutations recorded, the commonest being A382T (n = 11), G348C (n = 8), and M337V (n = 7). Five individuals with SOD1 mutation have bulbar onset, whereas 116 have limb onset (30 upper limb, 83 lower limb, three not specified). Twenty individuals with TARDBP mutation have bulbar onset, compared with 41 limb onset (10 upper limb, four lower limb, rest not specified). This difference in recorded presentation patterns between those with SOD1 and TARDBP mutations is highly significant (χ2 p-value 4 × 10−7).
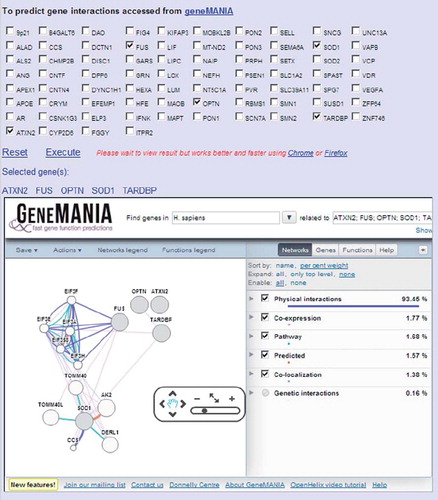
A GeneMANIA web interface allows prediction of ALS gene interactions in ALSoD, which is a powerful tool for the exploration of new pathways (Citation14) ().
Figure 4. Gene interactions using a GeneMANIA interface at ALSoD. Potential gene pathways and networks can be explored via the GeneMANIA feature on ALSoD, e.g. by including established or proposed ALS genes in the functional predictions.
Future implementations
ALSoD will widen its phenotypic remit to include frontotemporal dementia genes, develop more detailed geographical displays (for example, overlaying epidemiology study data on Google Earth), integrate genome-wide sequence data with appropriate display tools and, through collaborations, integrate transcriptomics data with existing gene and phenotype data. LOD scores and sequence data will be ported to a genome browser track for UCSC and Ensembl browsers allowing for easy analysis together with existing data as well as comparison with ALSGene results. Through collaboration with ALSGene, association evidence for phenotypic modifiers for age of onset, site of onset and survival will be displayed.
Assessing genetically complex forms of ALS: the ALSGene database
The methods to identify susceptibility variants for genetically complex traits differ from those aimed at finding new causative disease genes. Most commonly, susceptibility variants are identified by probing for significant differences in allele or genotype frequencies of a genetic variant across groups of affected (‘cases’) versus unaffected (‘controls’) individuals by performing genetic association analyses. As outlined in the introduction, several hundred such genetic association studies have been published in the field of sporadic ALS over the past two decades. As a result, more than 150 putative ALS risk genes have been reported. These findings have been met with inconclusive replication evidence, at best. More often than not, presumed risk effects were not observed in independent datasets, complicating an easy interpretation of these findings. More recently, genome-wide approaches have been applied to case-control datasets (Citation15–24) which – at least initially – did not lead to an improvement in replicability of the top findings. Concerted efforts using increasingly larger datasets have somewhat alleviated this situation (e.g. References 20,24). With a steadily growing number of GWAS and smaller-scale association studies focusing on sporadic ALS, the field becomes increasingly more difficult to evaluate. To facilitate this situation, our group has developed a systematic meta-analytic approach to genetic association studies that has already successfully been applied to other neurodegenerative diseases, e.g. Alzheimer's disease (Citation25) and Parkinson's disease (Citation26). We have begun applying the same systematic methodology to the field of sporadic ALS in the form of the ALSGene database.
Aims
ALSGene (freely available via the URL: http://www.alsgene.org or Prize4Life's ALS portal http://www.ResearchALS.org; see for a screenshot of the homepage of ALSGene) aims to serve as an exhaustive, unbiased, and regularly updated resource of genetic association studies in ALS. One of its key features will be up-to-date meta-analyses of all eligible genetic polymorphisms that have been investigated for association with ALS risk. This entails the inclusion of genome-wide association data, either in part or – if available – in full. All data and results are concisely summarized and displayed online. Upon completion of content curation, this will include an up-to-date list of ‘Top Results’, i.e. those showing the strongest evidence for association with risk for ALS when all available data are combined.
Figure 5. Screenshot of the ALSGene homepage (www.alsgene.org).
Methodology
For a visual summary of this paragraph see . Eligible studies are identified via PubMed searches, or by screening the table of contents of neurological and genetic journals. Additional publications are also sought by systematically screening the references of relevant publications (e.g. primary association papers, but also reviews, etc.). To date, these searches have yielded ∼500 articles on ALS genetics of which the full text is currently being screened for eligibility. A publication is included in ALSGene if it represents a genetic association study (here defined as studies investigating DNA sequence variants with ≥ 1% frequency in the general population, a.k.a. ‘polymorphisms’), and if it has been published in a peer-reviewed English language journal. Demographic details of included studies and genotype summary data of the investigated polymorphisms are extracted from each publication, and summarized on the ALSGene website. Each study included and listed in ALSGene is hyperlinked to the respective abstract in PubMed to facilitate identification and access to the original publication. In addition, an overview of all published ALS GWAS is provided in a dedicated section on ALSGene (http://www.alsgene.org/largescale.asp). Alongside the main characteristics of each study, this GWAS overview section also provides a hyperlinked list of ‘featured genes’, i.e. those loci or pathways highlighted by the primary authors as the main outcome of their study after having completed all analyses. ALSGene meta-analyses are performed for all genetic polymorphisms with at least four independent case-control datasets using allelic contrasts. Whenever possible, meta-analyses are complemented by GWAS data for overlapping polymorphisms (either directly genotyped or determined by imputation). Results of these meta-analyses are visualized on the website as forest plots (representing an up-to-date summary of the current evidence) and cumulative meta-analyses (summarizing the development of effect size estimates over time). Additional analyses include an assessment of the ‘epidemiologic credibility’ of significant findings using the HuGENet ‘Venice’ criteria (Citation27,Citation28), as well as Bayesian analyses (Citation29). Furthermore, meta-analyses are carried out after stratification for distinct ethnicities (e.g. Asian and Caucasian ethnicities), when at least three independent datasets for the respective ethnicity are available (for an example see ). More details on the methods behind data collection and analyses for ALSGene can be found online. All significant meta-analysis results are summarized in a dedicated section of ALSGene (‘Top Results’; note that the Top Results feature will only become available after primary literature-based content creation has been completed, i.e. approx. summer/fall of 2011).
Figure 6. Summary of ALSGene methodology. Flowchart of data selection, processing and analysis strategies applied for ALSGene. Polymorphisms with non-overlapping data from at least four data sets are subjected to meta-analyses. Significant meta-analysis results are highlighted in a designated section on the ALSGene homepage.

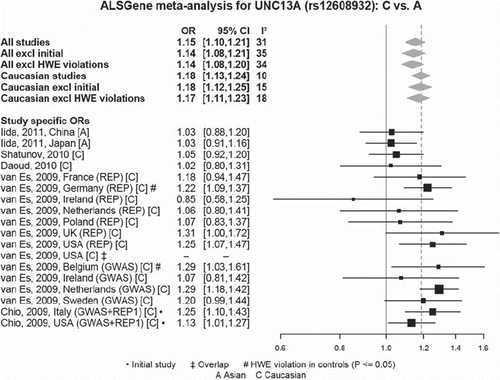
Figure 7. Random-effects meta-analysis of rs12608932 in UNC13A including all published studies (as of 1 March 2011). Example ALSGene forest plot providing a visual summary of meta-analysis results. Study-specific odds allelic ratios (ORs, bottom section) are displayed either as provided in the original publication or calculated de novo from allele or genotype summaries. Black squares represent point estimates of the OR (scaled to sample size), horizontal lines represent 95% confidence intervals. Summary ORs (top section) are then calculated from the study-specific ORs using random effects models for a number of different paradigms. Grey diamonds represent point estimates and 95% confidence intervals of the respective meta-analyses. I2 = a measure of between-study heterogeneity (see text for more details). Forest plots are only available for polymorphisms that have data available from four independent data sets.
Preliminary results
Currently, the online version of ALSGene displays the details and genotype summary data of over 90 independent studies that have investigated > 250 polymorphisms in 90 genes. As this paper is going to press, 21 meta-analyses are available, of which five show nominally significant results in four genetic loci (APEX1 (OR 0.78; p = 0.028), HFE (OR 1.72; p = 0.0034), UNC13A (OR 1.18 in Caucasians; p = 3.6 × 10−13; )), and a hitherto uncharacterized locus on chromosome 9p21.2 (OR 1.25; p = 5.3 × 10−18). The chromosome 9p21.2 locus and UNC13A, both initially implicated by recent GWAS (Citation20,Citation23,Citation24), currently show association with risk for ALS at genome-wide significance (i.e. p-values ≤ 5 × 10−8). The current UNC13A finding described here is a good example of why systematic data integration efforts are needed, even in the ‘GWAS era’ of complex genetics: as can be seen in , the four most recent individual datasets investigating the rs12608932 polymorphism in this gene only show very modest support in favour of a genuine risk effect (Citation24,Citation30,Citation31). In particular, the study by Iida et al. (Citation31) is interesting as this is the first to investigate this marker in non-European populations. At least in the investigated Chinese and Japanese datasets this polymorphism may have no or a much smaller role than in Europeans, although additional studies (e.g. with other markers in UNC13A) are needed to assess this more definitively. A similar situation of different effect size estimates across distinct ethnicities is observed for several of the current Top Results genes in our sister-database on Parkinson's disease genetics (PDGene, www.pdgene.org). Note that content for ALSGene is still under construction and that these results will probably change over time.
Future implementations
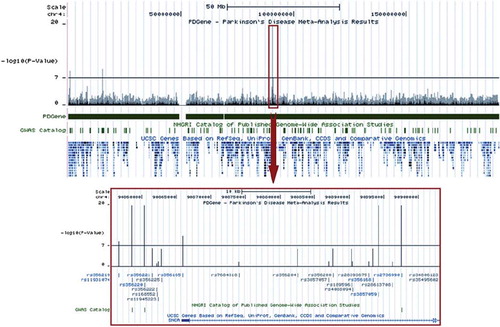
After completion of the initial curation process, all results will be ported to a customized UCSC genome-browser track displaying meta-analysis p-values. This will also include the results of meta-analyses on full GWAS datasets. This track will be hyperlinked to entries on ALSGene proper, where details of the sample-specific effect size estimates can be found for the most significant findings. shows an example screenshot of this feature for the PDGene database. In addition to displaying a browsable summary of ALSGene results on a genome-wide level, association signals can easily be put into the context of functional genetic studies, expression data, evolutionary conservation of genomic regions, etc., as annotated by the UCSC database (URL: http://genome.ucsc.edu/). This feature will also allow for an easy integration with content and results from the ALSoD database, as well as the comparison of genetic results emerging from our other database projects focusing on neurodegenerative disorders, such as Alzheimer's, Parkinson's, and frontotemporal dementia (FTDGene; in preparation).
Figure 8. Example screenshot of a customized UCSC genome-browser track (PDGene database) displaying meta-analysis p-values. The USCS genome-browser (http://genome.ucsc.edu/cgi-bin/hgGateway) presents an up-to-date, easy-to-navigate interface of the human genome. It integrates information from a broad variety of databases (e.g. gene-mapping, expression, evolutionary conservation) at base-pair resolution. Shown is an example screenshot for a customized browser track from the PDGene database maintained by our group (http://www.pdgene.org), which summarizes meta-analysis results (-log(10)) p-values, based on data from both genome-wide and candidate-gene association studies) equivalently at base-pair resolution. SNPs (rs-numbers) displayed in grey are based on meta-analyses of available GWAS datasets, those displayed in blue are supplemented by candidate-gene/replication datasets. All rs-numbers cross-link back to the PDGene database to provide more details on the respective meta-analyses. Note that this feature will become available on ALSGene once initial data entry and analysis is completed.
ALSoD and ALSGene: joining forces from two different angles
As outlined above, the respective focuses of ALSoD and ALSGene lie in different aspects of genetics research in ALS. The main focus for ALSoD is the summary and curation of genotype-phenotype correlations, which is usually clearest for Mendelian ALS genes and mutational studies, while ALSGene is mainly centred on genetic association studies. Both efforts face different challenges owing to the advent of powerful high-throughput technologies now allowing a growing number of research laboratories to routinely perform whole-genome genotyping and sequencing. In ALS genetics, two of the most interesting unanswered questions to date are: 1) are the same genes causing Mendelian forms of ALS through private mutations also involved in modifying disease susceptibility of non-Mendelian forms of ALS via the action of common polymorphisms (as seems to be the case in Parkinson's disease and frontotemporal dementia); and 2) can any of the genetic associations with common polymorphisms be explained by DNA variants that are neither common nor private? While these questions can only be directly answered by performing additional laboratory experiments, the outcomes of these efforts will be tracked and fed back to the research community by ALSoD and ALSGene. Combining the emerging evidence collected in both resources via an integrated interface (e.g. using the UCSC genome-browser architecture) is one efficient, yet simple, way of achieving this goal, in addition to hyperlinking related content and results across databases. In order to accomplish these objectives, the curatorial teams of both ALSoD and ALSGene have joined forces to provide a seamless and easy-to-navigate user experience, which will hopefully allow researchers to gain new insights from apparently unrelated sets of data.
Conclusions
With the advent of increasingly more powerful technologies, what we are learning about ALS is expanding every day. This is particularly true for research in human genetics, which has provided not only ground-breaking new insights into ALS pathogenesis, but also many insubstantial results. Online research databases such as ALSoD and ALSGene serve a vital purpose in facilitating access to knowledge and generating new insights from existing disparate sets of data allowing both genetics experts and non-experts to more easily separate the wheat from the chaff in the growing mountain of raw genetic data.
Acknowledgements
ALSoD is a joint project of the World Federation of Neurology (WFN) and European Network for the Cure ALS (ENCALS). We are especially grateful for the long-standing and continued funding of this project from the ALS Association and the MND Association of Great Britain and Northern Ireland. We also thank ALS Canada, MNDA Iceland and the ALS Therapy Alliance. The research leading to these results has received funding from the European Community's Health Seventh Framework Programme FP7/2007-2013) under grant agreement n° 259867. We also thank the NIHR Biomedical Research Centre for Mental Health. We thank Aleks Radunovic, Nigel Leigh, and Ian Gowrie, who originally conceived ALSoD, and the ALSoD team, especially Peter Andersen and John Powell.
ALSGene has been funded by Prize4Life as a critical functionality of their ALS Forum research portal (www.ResearchALS.org), created to serve as a unified source of breaking ALS research news and valuable tools for the ALS research community. It has been developed in close collaboration with the Alzheimer Research Forum. We thank Gabriele Strobel, Colin Knep and Paula Noyes for the online adaptation and maintenance of ALSGene. We further thank the ALSGene database team, especially Ute Zauft, Esther Meissner, Johannes Roehr, Brit-Maren Schjeide, and Leif Schjeide.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Schottkzy J. Ueber praesenile Verbloedungen. Z Ges Neurol Psychiat. 1932;140:333–97.
- Gowers WR. A Manual of Diseases of the Nervous System. Vol. I. Diseases of the nerves and spinal cord. 3rd. Philadelphia: P. Blakiston's Son & Co.; 1900.
- Aran FA. Recherches sur une maladie non encore decrite dy systeme musculaire (atrophie musculaire progressive). Archs Gen Med. 1850;3:5–35.
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, . Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
- Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online Mendelian Inheritance in Man (OMIM). Hum Mutat. 2000;15:57–61.
- Cotton RG, McKusick V, Scriver CR. The HUGO Mutation Database Initiative. Science. 1998;279:10–1.
- Scriver CR, Nowacki PM, Lehvaslaiho H. Guidelines and recommendations for content, structure, and deployment of mutation databases: II. Journey in progress. Hum Mutat. 2000;15:13–5.
- Claustres M, Horaitis O, Vanevski M, Cotton RG. Time for a unified system of mutation description and reporting: a review of locus-specific mutation databases. Genome Res. 2002;12:680–8.
- George RA, Smith TD, Callaghan S, Hardman L, Pierides C, Horaitis O, . General mutation databases: analysis and review. J Med Genet. 2008;45:65–70.
- Soussi T, Ishioka C, Claustres M, Beroud C. Locus-specific mutation databases: pitfalls and good practice based on the p53 experience. Nat Rev Cancer. 2006;6:83–90.
- Radunovic A, Leigh PN. ALSODatabase: database of SOD1 (and other) gene mutations in ALS on the internet. European FALS Group and ALSOD Consortium. Amyotroph Lateral Scler Other Motor Neuron Disord. 1999;1:45–9.
- Wroe R, Wai-Ling Butler A, Andersen PM, Powell JF, Al-Chalabi A. ALSOD: the Amyotrophic Lateral Sclerosis Online Database. Amyotroph Lateral Scler. 2008;9:249–50.
- Yoshida M, Takahashi Y, Koike A, Fukuda Y, Goto J, Tsuji S. A mutation database for amyotrophic lateral sclerosis. Hum Mutat. 2010;31:1003–10.
- Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, . The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38: 214–20.
- Dunckley T, Huentelman MJ, Craig DW, Pearson JV, Szelinger S, Joshipura K, . Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med. 2007; 357:775–88.
- Schymick JC, Scholz SW, Fung HC, Britton A, Arepalli S, Gibbs JR, . Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2007;6:322–8.
- van Es MA, van Vught PW, Blauw HM, Franke L, Saris CG, Andersen PM, . ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 2007;6:869–77.
- Cronin S, Berger S, Ding J, Schymick JC, Washecka N, Hernandez DG, . A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet. 2008;17:768–74.
- van Es MA, van Vught PW, Blauw HM, Franke L, Saris CG, van den Bosch L, . Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet. 2008;40:29–31.
- van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, . Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41: 1083–7.
- Landers JE, Melki J, Meininger V, Glass JD, van den Berg LH, van Es MA, . Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009;106:9004–9.
- Chio A, Schymick JC, Restagno G, Scholz SW, Lombardo F, Lai SL, . A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:1524–32.
- Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai SL, Myllykangas L, . Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–85.
- Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, . Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9: 986–94.
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer's disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23.
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, . The PDGene Database. Alzheimer Research Forum. Available at: http://www.pdgene.org/.
- Ioannidis JP, Boffetta P, Little J, O'Brien TR, Uitterlinden AG, Vineis P, . Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol. 2008;37:120–32.
- Khoury MJ, Bertram L, Boffetta P, Butterworth AS, Chanock SJ, Dolan SM, . Genome-wide association studies, field synopses, and the development of the knowledge base on genetic variation and human diseases. Am J Epidemiol. 2009;170:269–79.
- Ioannidis JP. Effect of formal statistical significance on the credibility of observational associations. Am J Epidemiol. 2008;168:374–83; discussion 384–90.
- Daoud H, Belzil V, Desjarlais A, Camu W, Dion PA, Rouleau GA. Analysis of the UNC13A gene as a risk factor for sporadic amyotrophic lateral sclerosis. Arch Neurol. 2010;67: 516–7.
- Iida A, Takahashi A, Deng M, Zhang Y, Wang J, Atsuta N, . Replication analysis of SNPs on 9p21.2 and 19p13.3 with amyotrophic lateral sclerosis in East Asians. Neurobiol Aging. 2011;32:757. e13–4.