
ABSTRACT
Endometriosis (EM) and uterine leiomyoma (UL) are two most frequent benign tumors of monoclonal origin affecting about 30% of all women in their reproductive age. Modern molecular technologies have made a tremendous impact in understanding both disorders. Here is the first comparative analysis of molecular mechanisms underlying development of EM and UL as it looks from the platform of systems genetics. Similarities and differences of EM and UL at their incipient stages are enlightened with special emphasis on their gene networks, gene expression, and epigenetic regulation, of pathologic development. The analysis substantiates a new hypothesis postulating tumors as outgrowths of the stem cells with mesenchymal commitment lineage (mSC) which migrate from the endometrium/myometrium junctional zone of the uterus. Comparative analysis has revealed basic similarities of molecular pathogenesis of EM and UL suggesting molecular syntropy of both disorders. Peculiarities of the epigenetic landscape determining development of mSC may explain the existence of different clinical forms of EM and UL as well as their unique clinical manifestation. Some perspectives for practical and scientific application in EM and UL studies of this new hypothesis are outlined.
Abbreviations
AM: adenomyosis; CGH: comparative genomic hybridization; ECM: extracellular matrix; ESP: endometrial/ progenitor stromal stem cells; EM: endometriosis; EML: endometriotic lesions; EMT: epithelieal–mesenchymal transition; ER: estrogen receptor; JZ: junctional zone; FGF: fibroblast growth factor; FH: fumarate hydrolase; GWAS: genome-wide association studies; HMGAs: high mobility group proteins; LM: leiomyoma; LMSP: leiomyoma-derived side population; MED12: mediator subcomplex 12; NGS: new generation sequencing; PR: progesterone receptor; SC: stem cell; mSC: stem cells with mesnchymal lineage commitment; SMCs: smooth muscle cells; STAT: signal transducer and activator of transcription; UL: uterine leiomyoma
Introduction
Endometriosis (EM) and uterine leiomyoma (UL) are common complex disorders, affecting over 30% of all women in their reproductive age (6-10% for EM and 20-30% for UL) [Burney and Giudice Citation2012; Okolo Citation2008]. Both are known as benign tumors of the female genital tract with EM stemming from cycling endometrium and UL from smooth muscle of the myometrium and have many traits in common (). Our recent review concerned new options using epigenetic approaches for understanding the development mechanisms and clinical manifestation peculiarities in EM [Baranov et al. Citation2015].
Table 1. Basic clinical and genetic traits of endometriosis and uterine leiomyoma.
To our knowledge this article is the first attempt of comparative analysis of molecular mechanisms operative in the development of EM and leiomyoma (LM) starting from the origin and tumor drivers through progression and peculiarities of manifestation. The analysis supports the view of the existence of molecular similarity (syntropy) of both disorders and substantiates some developmental hypotheses. Stem cells with mesenchymal commitment (mSC) within a junctional zone (JZ) of the uterus are the most plausible candidate cells giving rise to both tumors. Different clinical forms and manifestation peculiarities of EM and UL stem from deviations in epigenetic landscape routes chosen by migrating mSC after their tumor transformation.
Somatic stem cells (SCs) are a subset of cells residing in normal adult tissues that, through asymmetric division, retain their ability to self-renew while producing daughter cells that go on to differentiate and play a role in regeneration and repair of the native host tissue [Ono et al. Citation2012; Schwab et al. Citation2008]. Thus their differentiation capacities are usually restricted to the tissue’s derivatives of particular germ layer of their origin.
Origin of EM
A unifying theory regarding the origin of EM remains elusive so far. Tentatively, it could be attributed either to implantation in peritoneum of otherwise normal cells of endometrum from menstrual debris or to metaplasia of preexisting undifferentiated epithelium cells from peritoneum [Baranov et al. Citation2015; Signorilea and Baldia Citation2015]. Since 1991 it has been suggested that the high regenerative capacity of human endometrium is mediated by SCs [Padykula Citation1991]. It was later shown that specific SC markers such as C-kit (CD117) and CD34 are expressed in progenitor and stromal cells of the endometrium [Chan et al. Citation2004]. The expression of other well-known SC markers, e.g., OCT4 and NANOG is significantly higher in ectopic endometriotic tissues, compared with normal endometrium or myometrium [Chang et al. Citation2013]. The level of OCT4 messenger RNA in endometriotic tissues is positively correlated with the expression of the genes associated with cell migration. Endometrial SCs have been also shown to present the CD146, PDGF-Rb and CD146, CD90 high cell surface markers [Maruyama Citation2014]. The identification of SC markers confirms the presence of endometrial progenitor/stem cells (ESP) population in EM lesions (EML), thus supporting ‘stem cell theory’ of EM [Sotomayor et al. Citation2009].
Concerning the location of ESPs in the uterus it should be noted that normally endometrium is composed of two layers. The outer functional or cycling layer, contains epithelial and stromal cells which are shed monthly under the influence of circulating estrogens and progesterone [Sasson and Taylor Citation2008; Su et al. Citation2010]. The inner basal layer, resting on the myometrium, close to the myometrium/endometrium JZ, serves to rebuild a new functional compartment and thus is considered a preferable site for ESPs [Kalluri Citation2009]. EML cells arise from translocated endometrial stem/progenitor cells [Huang et al. Citation2011]. They contain self-renewing mSCs expressesing SC markers and demonstrate a monoclonal nature in individual glands [Gotte et al. Citation2008].
There is some evidence in favor of an ESP origin of EM [Forte et al. Citation2014]. First, they are found within the functional layer of tissue that is translocated during retrograde menstruation (implantation theory). Second, they can attach, migrate, and initiate angiogenesis at an ectopic site [Pittatore et al. 2013]. Finally, the cells possess the capability to differentiate into multiple lineages to reconstitute the endometrium at an ectopic site, to stimulate endometrial epithelial and stromal cells in vitro and in vivo, to undergo mesenchymal-to-epithelial transition (MET) and vise-verse (EMT) [Maruyama Citation2014].
It is important to stress that ESPs are multipotent, as their progeny in vitro can differentiate into adipocytes, chondrocytes, osteoblasts, and even smooth muscle cells (SMCs) [Gargett et al. Citation2009]. The latter are frequently found in peritoneal, ovarian, and deep-infiltrating EMLs [Aznaurova et al. Citation2014]. The origin of SMCs within EML remains unknown but looks very intriguing and advocates in favor of common molecular mechanisms underlying EM and UL.
Origin of UL
The cell population of myometrium giving rise to UL has been identified not long ago. Support for the existence of SCs as a side-population in human myometrium has been obtained from a series of experiments with 5-bromo-2 -deoxyuridine (BrdU) [Ono et al. Citation2012]. Recently, it has been reported that side population cells are present in UL [Mas et al. Citation2014]. Isolated in vitro, the undifferentiated status of side population cells from UL has been confirmed by the expression of OCT4, NANOG, DNMT3B, and GDF3 genes and by high proliferative capacity under hypoxic conditions in vitro [Ciavattini et al. Citation2013]. Mesenchymal lineage commitment of these cells was demonstrated by their ability to differentiate in vitro into adipogenic and osteogenic lineages. A distinct stem cell-enriched population, designated as the UL-derived side population (ULSP), is responsible for cell proliferation and tumor growth. It comprises approximately 1% of all UL cells and 2% of all myometrium-derived cells. ULSPs arise from myometrial cell transformation, though its timing has not been elucidated so far and may occur in adults or during an embryonic stage. It is worth noting that ULSP cells are predominantly localized in the myometrium part of the JZ close to the endometrium layer [Tocci et al. Citation2008]. The cells of JZ exhibit significant differences compared with those of other myometrium [Sun et al. Citation2015]. Abnormal thickening of the JZ is considered the causative factor in the development of adenomyosis (ADS)-outgrowth of a benign endometrial tumor (similar to EM) within the myometrium [Zhang et al. Citation2015].
Thus it might be inferred that mSCs are preferably concentrated in the JZ of the uterus and possess high regenerative capacities necessary for remodeling and cycling of endometrium and myometrium tissues. It also looks plausible that both UL and EM tumors are coming from the mSCs which infest myometrium/endometrium JZ and possess mesenchymal lineage commitment. Proximity in their place of origin, identical or very similar membrane markers, and mesenchymal lineage make mSCs very intriguing candidates as the source and molecular pathogenesis of EM and UL. Tumor transformation of endometrium and smooth muscle cells is still poorly understood. Molecular, biochemical, and cytogenetic mechanisms of this metaplasia remain the area of intensive studies.
Initiation of tumorigenicity: EM
The contribution of the mesenchymal–epithelial and epithelial–mesenchymal transition (EMT) processes in the pathogenesis of pelvic EM has been repeatedly shown [Matsuzaki and Darcha Citation2012; Forte et al Citation2014]. Actually, according to recent data [Proestling et al. Citation2015] transformation to ectopic EML starts with the transition of epithelial cells of the endometrium into stromal mesenchymal cells (EMT). Epithelial cells lose their polarity, cell-to-cell contacts, and acquire migratory and invasive abilities of mesenchymal cells. The latter are inevitable for establishment of endometrium defects encountered in EML. Molecular mechanisms underlying EMT involve the functional loss of E-cadherin (CDH1) in epithelial cells induced by transcription factor TWIST1 [Yang et al. Citation2004]. The latter is significantly overexpressed in ectopic EML compared to normal endometrium [Thiery et al. Citation2009].
Expression of the anti-apoptotic factor BCL-2 and reduction of the pro-apoptotic factor BAX in EML provides a survival mechanism of EM cells in ectopic sites [Beliard et al. Citation2004]. In contrast, increased expression of genes involved in cell cycle progression, such as the overexpression of MYC and Cyclin D1, provide a higher cell proliferation rate in EML [Pellegrini et al. Citation2012]. Several studies investigating MYC expression observed increased MYC mRNA and protein expression in ectopic and eutopic endometrium from EM patients [Proestling et al. Citation2015]. Thus it might be inferred from this study that transcription factor TWIST1 is an important regulator of EMT in endometrium and obviously upregulates the onset of ectopic EML. Taking into account the great heterogeneity of EM lesions [Proestling et al. Citation2015], EMT with subsequent metaplasia into tumor cells might be considered as a rather important pathway to tumorigenicity in EM.
Chromosome aberrations and epigenetic deregulation might also contribute to tumor transformation in EM. Karyotype anomalies, consistent with chromosome instability have been confirmed in EM cells by array-CGH and fluorescence in situ hybridization (FISH) studies. Array-CGH analysis of DNA from patients with ovarian EM has revealed a genomic duplication of the chromosomal region 20q13.33 with concomitant overexpression of GATA5 (transcription factor of GATA-binding motif) and SLCO4A1 (solute carrier organic anion transporter family member 4A1) genes in approximately 50% of the patients [Yang et al. Citation2013]. Some of the submicroscopic aberrations close to oncogenes and tumor suppressor genes may gain the drivers function of EM [Jolinière et al. Citation2014]. Loss of chromosome 17 centromere as well as oncosuppressor p53 locus regions (17p13.1) has also been registered in specimens from advanced EM [Bischoff et al. Citation2002].
Initiation of tumorigenicity: UL
Molecular mechanisms underlying tumorigenicity of smooth muscle cells remain rather obscure. It is known that both normal myometrium and fibroid tissue contain pools of cells with the capacity for self-renewal, which are referred to as SCs. The latter are responsible for the proliferation of normal myometrial SCs used for the physiologic enlargement of the uterus during pregnancy. Mature smooth muscle cells (SMC) express much higher levels of estrogen receptor α (ERα) and progesterone receptor (PR) than do SCs of smooth muscle. It is likely therefore that estrogen- and progesterone-dependent cell proliferation is primarily mediated by the ERα and PR that reside in mature cells. Paracrine factors, such as WNT ligands, that are released by mature cells may act on SCs to induce their self-renewal and proliferation [Bulun Citation2013]. However the exact nature of cell population that gives rise to UL is not known [Laughlin et al. Citation2010].
It should be recognized that genetic abnormalities in SCs play pivotal roles in the tumorigenesis of smooth muscle SCs [Ono et al. Citation2012]. Recent findings on genetic changes of UL suggest that these benign tumors are a heterogeneous group of diseases in terms of their molecular pathogenesis with genetic and chromosomal abnormalities as principal tumor-initiating factors. Almost 25% of UL possess visible karyotype alterations including translocations, chromosome imbalances, and complex rearrangements caused by chromotrypsis [Holzmann et al. Citation2014]. The major part of UL (70-75%) is characterized by mutations of mediator subcomplex 12 gene MED12 and just a few (1-1.5%) by mutations of the FH (fumarate hydrolase) gene. Submicroscopic alterations detected by CGH are also not rare [Markowski et al. Citation2011; Jolinière et al. Citation2014]. Chromosome aberrations and rearrangements in UL cells are often close to the genes of high mobility group proteins (HMGAs) and DNA repair (RAD51B).
As the next step point mutations and chromosome aberrations may activate signal transduction pathways such as Wnt/β-catenin or Wnt/MAPK [Mas et al. Citation2014] which stimulate SCs proliferation and transformation into self-renewed UL SCs, which divide in an uncontrolled fashion, give rise to mature fibroids. It is also important to stress that the WNT gene, acting through the frizzled family of receptors, activates the β-catenin–T-cell transcription factor (TCF) pathway, which induces overexpression of transforming growth factor β (TGF-β) and leads to excessive formation of extracellular matrix [Bulun Citation2013]. The absence of MED12 or the presence of its mutant form in SCs has also been linked to increased expression of the TGF-β receptor, which leads to the activation of its downstream signaling including mitogen-activated protein kinase (MAPK) family proteins, mediating stem-cell self-renewal and proliferation [Ono et al. Citation2012].
It is widely accepted that tumorigenic transformation of SCs can be provoked by many unfavorable conditions, including muscle contractions at menstruation and during labor [Moroni et al. Citation2015]. The non-random location of primary UL nodules within the myometrium is repeatedly reported [Ciavattini et al. Citation2013]. Of particular interest is their permanent position within irregular thickenings of JZ of myometrium, which is considered a regular site of origin for submucosal and intramural UL fibroids [Larsen et al. Citation2011].
As might be inferred, though still hypothetical, initial tumorigenicity mechanisms of SCs progression giving rise to EM and UL reveals many features in common. The latter include activation of antiapoptotic and mitogenic pathways, changes of inter cell communication, storage of excessive extracellular matrix, and increase in cell migration. Some differences in early tumorigenicity of EM and UL are also quite evident. While EM starts from epigenetically governed epithelial cells transition into mesenchymal cells (EMT) accompanied with some minor non-specific chromosomal aberrations, mutations of MED-12 in conjunction with some specific chromosomal rearrangements involving HMGAs genes are treated as primary tumor drivers in UL development.
Gene interactions
Complex gene networks of EM and UM are shown in . It should be noted at their start, both disorders have almost identical functional groups of candidate genes. That is, genes regulating proliferation and embryogenesis, tumor suppressors and oncogenes, growth factors and angiogenesis, cell matrix and cell adhesion, hormones and their receptors, cytokines and their receptors, as well as miRNA synthesis. A detoxification group of genes identified by candidate gene studies in EM is the only exception, as association studies identified candidate genes for EM but not UL. With minor exception (see below) most of these genes were identified in functional mapping and thus their association needs further ascertainment [Zondervan et al. Citation2002]. Nonetheless sharing the same functional groups of the genes a priori suggests molecular proximity of EM and UL. It should also be emphasized that just a few candidate genes are shared by each of six functional groups in EM and UL while the vast majority of already identified or suspected candidate gene moieties are different thus indicating the specificity of molecular mechanisms underlying progression of both disorders ().
Figure 1. Epigenetic landscape and gene network of endometriosis (EM) and uterine leiomyoma (UL). (Top center) ‘Epigenetic landscape’ of mesenchymal stem cell (mSC) development resulting in EM or UL. ‘Epigenetic landscape’ shows branching channels (paths) - feasible ways taken by differentiating mSC. After the onset of pathological process provoked by some alien intrinsic and extrinsic factors mSC undergo blast transformation and proceed further development with many variations resulting in different clinical manifestations of the diseases. (Remaining columns) Main functional groups of the gene-networks associated with the origin and development of EM and UL. Meaningful associated genes are in bolds. The rest are candidate genes identified in functional mapping studies [Rahmioglu et al. Citation2014; Baranov et al. Citation2015].
![Figure 1. Epigenetic landscape and gene network of endometriosis (EM) and uterine leiomyoma (UL). (Top center) ‘Epigenetic landscape’ of mesenchymal stem cell (mSC) development resulting in EM or UL. ‘Epigenetic landscape’ shows branching channels (paths) - feasible ways taken by differentiating mSC. After the onset of pathological process provoked by some alien intrinsic and extrinsic factors mSC undergo blast transformation and proceed further development with many variations resulting in different clinical manifestations of the diseases. (Remaining columns) Main functional groups of the gene-networks associated with the origin and development of EM and UL. Meaningful associated genes are in bolds. The rest are candidate genes identified in functional mapping studies [Rahmioglu et al. Citation2014; Baranov et al. Citation2015].](/cms/asset/9b227e5b-08b6-4c34-9434-39523e77058f/iaan_a_1123325_f0001_oc.jpg)
Gene interactions: EM
Candidate gene association studies, whole genome linkage analysis, as well as, more recent, genome-wide association studies (GWAS) were applied and resulted in the identification of over 100 candidate genes associated with EM [Baranov et al. Citation2015]. Most were identified by functional mapping analysis with rather variable results. Their significance for understanding molecular pathogenesis of EM remains problematic. Meanwhile, some EM candidate genes (, bold letters) mapped by QTL analysis and identified in meta analysis of 8 GWAS carried out with a unprecedentedly high number of EM patients (over 11,000) are now considered as meaningful entities of EM [Rahimyoglu et al. Citation2014]. A highly reliable association was shown for 6 genes: WNT4, VEZT, CDKN2b-AS1, ID4, GREB, and NFE2L3. More distant candidate genes were HOXA, HOXA10, and FN1. One gene, CYP2C19 (aromatase) has been identified in earlier linkage studies [Rahimyoglu et al. Citation2014].
Total genetic impact in EM was estimated around 52%, and 8 of 9 loci were likely to be implicated predominantly in the development of moderate/severe forms of EM (stages III/IV American Society for Reproductive Medicine (1996) [Canis et al. Citation1997]). One of the candidate genes, WNT4 is of special interest for pathogenesis of EM as it plays decisive roles during the female reproductive tract development. The genes of the WNT family are expressed to endometrium, suggesting that EM can also arise through metaplasia of normal peritoneum cells [Kobayashi et al. Citation2014; Signorilea and Baldia Citation2015]. One more candidate gene closely associated with EM is GREB1 (2p25.1), participating in estrogen regulating pathway, including estrogen stimulated cell proliferation. Increased expression in EM lesions makes its involvement in molecular pathogenesis of EM very plausible [Fung et al. Citation2015].
Protein products fibronectin and vezatin of two other candidate genes (FN1 and VEZT, respectively) resolved from the meta-analysis are responsible for cell adhesion, migration, and transmembrane cell junction, thus being quite in line with the hypothesis of epithelieal–mesenchymal transition (EMT) [Forte et al. Citation2014; Proestling et al. Citation2015]. Ovarian oncogene ID4 and tumor suppressor gene CDKN2B-AS1 have also been implicated in molecular pathogenesis of EM either as part of the regulation of the HOXA9 and CDKN1A transcriptional program (ID4) or as an oncosupressor inactivated by hypermethylation of its promoter (CDKN2B-AS1). However, assuming a population risk for EM of about 8% Zondervan et al. [Citation2001; Citation2002], identified nine loci that only account for 1.67% of all variance in EM susceptibility; thus predictive genetic testing of EM is still problematic.
Although no specific mutations or chromosomal aberrations are yet known for EM, each functional group involved in its pathogenesis has at least one or more genes that link with EM in a meaningful manner. As follows from , most of these genes are responsible for cell proliferation, control embryonic development, or are involved in tumorigenesis. Additional detail of the other candidate gene groups in EM pathogenesis is provided elsewhere [Baranov et al. Citation2015].
Gene interactions: UL
Contrary to EM, specific chromosome aberrations and mutations of at least two genes MED12 and FH are known as principal ‘drivers’ of UL. The most common MED12 gene mutation is encountered in about 70% of UL fibroids [Mäkinen et al. Citation2013]. The MED12 gene product is known as a part of a huge protein complex (the Mediator) with intermediate functions between RNA polymerase II and transcription factors. MED12 connects cyclin C-CDK8 to the core of the mediator complex and is required for kinase activity of the CDK8. This regulates the expression of many genes including TP53, WNT, SHH, and other signaling pathways [Wei et al. Citation2005]. MED12 is implicated in transcription activation of Wnt target genes by interacting with β-catenin [Kim et al. Citation2006]. Another UL candidate is the HMGA2 gene mapped close to break points in translocation 12:14, the most common UL cytogenetic finding, encountered in about 20% of chromosomally abnormal UL lesions. This gene encodes a high mobility group DNA binding protein and embryonic proliferation modulator [Ciavattini et al. Citation2013; Markowski et al. Citation2011]. ULs that do not carry a cytogenetic aberration overexpress the HMGA2 gene compared with myometrium, suggesting that HMGA2 plays an important role in UL development even in those fibroids which do not carry the specific t(12;14) rearrangement [Holzmann et al. Citation2014].
Recent data suggest that both pathways may account for a considerable percentage of UL rearrangements of high mobility group protein gene 2 (HMGA2) and, much more frequently, those affecting MED12. Apparently, both mutations do not overlap with each other [Bertsch et al. Citation2014] and seem to be associated with different tumor sizes [Ono et al. Citation2012] as well as with their presence as multiple versus solitary tumors [Markowski et al. Citation2014; Osinovskaya et al. Citation2015]. Tumors with HMGA2-rearrangements are less frequent than their MED12-mutated counterparts [Makinen et al. Citation2013] and preferentially occur in larger, solitary lesions [Holzmann et al. Citation2014]. Moreover, in contrast to UL cells harboring MED12 mutations, the cells with HMGA2-rearrangements, are able to survive in vitro for numerous passages [Bulun Citation2013]. In a considerable percentage of UL (approximately 15%) neither chromosomal, no gene mutations are found. They include cases with rare driver mutations such as deletions of the FH-locus, chromotrypsis and other types of submicroscopic imbalance, detected by CGH [Holzmann et al. Citation2014]. The occurrence of gene mutations in UL has been suspected to be much higher than that of chromosome aberrations [Bertsch et al. Citation2014].
As might be inferred from , many other genes are involved in molecular mechanisms of UL development. These include the imbalance of steroid sex hormones, dysregulation of microRNA, growth factors, cytokines, chemokines, and defects of extracellular matrix components and its receptors PR-A and PR-B. They all play key roles in UL biology [Kim and Sefton Citation2012; Bulun Citation2013]. Progesterone content and its mRNA level are higher in UL than those in normal myometrium. Overexpression of PR-B mRNA at the surface of UM upregulates the EGF gene (mitogenic) [Ying and Weiyuan Citation2009] and transforming growth factor gene TGF-β3 [Fujimoto et al. Citation1998]. PR is essential for the response of fibroid tissue to progesterone secreted by the ovaries. Progesterone and PR are indispensible to tumor growth, increasing cell proliferation and survival, and enhancing extracellular-matrix formation.
Estrogen responsive genes mRNA and protein expression levels as well as the content of ER-α and ER-β are higher in UL compared to those in normal myometrium [Benassayag et al. Citation1999]. Estrogens may exert UL growth by stimulatory effects on cytokines, growth factors, or apoptosis factors [Grings et al. Citation2012] by suppressing normal p53 functions. Estrogens are able to regulate the expression of growth factors by activating some signaling pathways. Growth factor genes (EGF, HB-EGF, VEGF, bFGF,PDGF, TGF-b, and ADM) are differentially expressed in LM compared with normal myometrium [Tal and Segars Citation2014]. VEGF, EGF, heparin binding epidermal growth factor (HB-EGF), PDGF, IGF, TGF- α, TGF-β, acidic fibroblast growth factor (aFGF), and basic fibroblast growth factor (bFGF), and their receptors have been demonstrated to play a crucial role in UL growth [Norian et al. Citation2009; Malik and Catherino Citation2012]. Some growth factors are known as inducers of mitogenesis and differentiation of mesodermal cells, including fibroblasts, stem myoblast cells (SMCs), and endothelial cells. Several angiogenic genes show consistent differential expression in UL compared with normal myometrium. Namely, TGF-b is up-regulated, while connective tissue growth factor (CTGF) and cysteine-rich angiogenic inducer 61 (CYR61) are steadily down-regulated in UL if compared with normal myometrium [Leppert et al. Citation2006; Tal and Segars Citation2014].
Many cytokines, chemokines, their receptors, interleukin-(IL-)1, IL-6, as well as tumor necrosis factor-β and erythropoietin have been suspected in UL development [Ciavattini et al. Citation2013]. ECM microenvironment of UL cells is characterized by increased mechanical stress and suggests that the mechanical properties of UL are a key feature of these tumors and may contribute to their growth [Norian et al. Citation2012]. Decisive changes potentiating UL growth occur in extracellular matrix (ECM). Pathogenesis of fibroids seems to involve a positive feedback loop between extracellular matrix production and cell proliferation and vitamin D. Extracellular matrix components mainly consisting of collagen subtypes, fibronectin, and proteoglycans, a series of collagen subtypes, such as COL1A1, 4A2, 6A1, 6A2, 7A1, and 16A1 have been recently found expressed to a greater extent in UL cells compared to normal myometrial cells [Malik et al. Citation2010].
Epigenetic regulation of EM
Common epigenetic mechanisms of regulating gene activity such as DNA methylation or specific micro-RNAs are known both for EM and UL. Dysregulation of methylation in EM tissues is especially obvious for the genes involved in sex-hormone metabolism, cell proliferation, and embryonic development (tumor suppression, immunological effects, vessel growth, and cell interactions) [Baranov et al. Citation2015]. Typical for EM is hypomethylation and thus up-regulation of estrogen receptor-beta (ESR2), aromatase (CYP19A1) gene, and steroidogenic factor-1 (NR5A1) while hypermethylation (down-regulation) was predominantly confined to promoters regions of progesterone receptor (PGR)-B, histone deacetylase inhibitors (HDACi), and methyltransferases (DNMT) genes [Wang et al. Citation2007].
Comparative analysis of methylation profiles in normal endometrium and in EM cells reported obvious methylation differences in 403 genes, predominantly transcription factors. [Houshdaran et al. Citation2014]. Shift in methylation of two isoforms of transcription factor GATA (GATA2 and GATA6) results in impairment of hormonal sensitivity and aberrant expression of many candidate EM genes [Dyson et al. Citation2014]. Hypomethylation in the promoter region of COX-2 increases the production of prostoglandines (PGF2)-major contributors to severe endometriosis-related dysmenorrheal and inflammation [Wang et al. Citation2007]. Impaired methylation and thus expression has also been reported for many imprinted genes of EM [Kobayashi Citation2014].
Another important epigenetic mechanism is miRNA regulation. Given the diverse roles that miRNAs play in cellular functions it is not surprising that they play key regulatory roles in the pathogenesisis of EM. Numerous miRNAs are mis-expressed in EM cells and abnormal profiles of miRNAs are reported [Suryawanshi et al. Citation2013]. Some of these miRNA are definitely over- or under-expressed in EM tissues. But none of these miRNAs show statistically meaningful deviations from the control. Moreover miRNAs levels in EM tissue are not correlated with serum or plasma miRNAs from women with EM and thus they cannot serve as relatively non-invasive biomarkers of EM [Nothnick et al. Citation2015].
At least some known miRNAs deserve special attention. One of them is miRNA-148a [Rahamiglou et al. Citation2014]. It participates in the regulation of WNT/β-catenin signaling pathway which provides communication between epithelial and stromal cells in endometrium [Tulac et al. Citation2003]. This pathway, so important for cell proliferation, migration, and invasion, might be directly involved in EML development. In addition, miR-196b is significantly repressed in EM cells though it demonstrates anti-proliferative and pro-apoptotic functions by targeting c-myc and Bcl-2 genes expression. Another example includes miR-210; through signal transduction and targeted activation of STAT3 signaling pathway it promotes many important cell reactions in EM lesions including proliferation, resistance to apoptosis, and angiogenesis [Okamoto et al. Citation2014]. Moreover it has been demonstrated that miR-210 induces normal endometrial cells to differentiate into the endometriotic phenotype. It was speculated that up-regulated miR-210 expression in endometrial stromal/stem cells is involved in the creation of endometriosis specific cellular dysfunction through epigenetic mechanisms. This yielded either activation or inactivation of numerous products in the STAT3 signaling pathway. It has also been suggested that STAT3 inhibitors should be tried as potential candidates for the treatment of EM [Montgomery et al. Citation2014].
Epigenetic regulation of UL
Epigenetic mechanisms including DNA methylation, histone modification, and microRNAs in UL are actively studied and provide an important clue to the origin of this tumor [Mas et al. Citation2014]. Follow-up studies demonstrate abnormal global genomic methylation in UL compared to normal myometrium [Yamagata et al. Citation2009], implicating possible epigenetic contributions to genetic susceptibility of UL development. Several miRNAs such as let7, miR-21, miR-93, miR-106b, and miR-200 are significantly deregulated in UL cells compared to normal myometrium [Marsh et al. Citation2008]. Growing evidence indicates that steroid hormones E2 and P are the most important epigenetic regulators of growth factors, cytokines, chemokines, and extracellular matrix components that are so important for UL progression. Epigenetic changes could activate signal transduction pathways such as Wnt/ β-catenin or Wnt/MAPK [Bulun Citation2013; Ciavattini et al. Citation2013; Tal and Segars Citation2014].
Thus all basic mechanisms of epigenetic control such as DNA methylation, histone modification, and miRNA synthesis are dysregulated in EM and UL. However, the details of impaired epigenetic regulation seem to be different and rather specific for each disorder. The differences concern both methylation profile specificity and miRNA patterns. Not a single of more than 20 already identified miRNAs that was significantly dysregulated in EM and UL was found in common for both pathologies so far.
EM and UL as syntropic disorders
Initially the term ‘syntropy’ was coined to designate the diseases that tend to occur in patients or their relatives more often than what is expected by chance [Pfaundler and von Sehr Citation1922. Now at the age of molecular medicine syntropy is usually treated as a natural genetic phenomenon of nonrandom combination of two or more genetically related pathological configurations affecting the same or several individuals and their relatives [Puzyrev et al. Citation2010]. Autoimmune diseases, cardiovascular or psychological diseases are well known examples of syntropy.
As might be inferred from previous sections and , EM and UL fit basic demands of syntropic diseases. Their pathogenesis and pathophysiology share many molecular mechanisms in common. Both are hormonal dependent benign tumors of monoclonal origin coming from uterine tissues. They are not rare in the same patient. Such an association reaches up to 60% for intrinsic form of EM - adenomyosis (AM) (emdometrial outgrowth into myometrium) and about 70% of AM patients also demonstrate external genital EM [Kunz et al. Citation2005].
The crucial argument in favor of EM and UL syntropy can be addressed as the identity or obvious similarity of their progenitor/stem cells (). A growing bulk of evidence suggests both tumors start from the relevant progenitor/stem cells with mSCs (see origin of EM and UL and initiation of tumorigenicity of EM and UL sections discussed above). The latter is shown by their ability to differentiate into adipogenic, osteogenic, or chondrogenic lineages [Gargett et al. Citation2009; Ciavattini et al. Citation2013]. It is also suspected that EM lesions are present from an early age or can be congenitally acquired and their development occur after menarche due to the sex steroid hormones’ stimulation [Signorilea and Baldia Citation2015]. This plausible hypothesis requires further support.
Figure 2. Junctional zone (JZ) of the uterus as ‘the same coin’. Myometrial/endometrial JZ of the uterus at the boundary of endometrium/myometrium layers as a principal source of mesenchymal stem cells (mSC), generating under unfavorable conditions endometriosis (EM) lesions or uterine leiomyoma (UL) fibroids. The mSC from the upper (myometrial) side of the ‘coin’ are epigenetically programmed to generate UL, and the mSC from the bottom (endometrial) side are prone to give EM lesions.
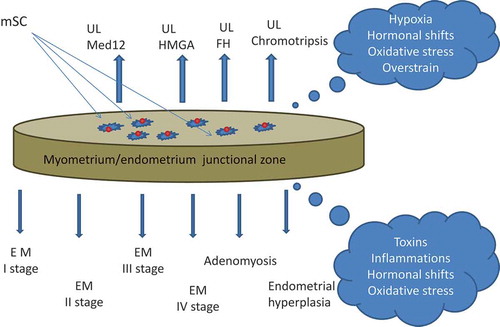
The close relationship of EM and UL is also favored by sharing a common site of the origin. Cambial JZ at the endometrium/myometrium boundary should be considered the most suitable cradle for these mSCs. Two lines of evidence support this notion. As it was mentioned above, the subendometrial myometrium is a site of origin of submucosal and intramural fibroids and the progeny of SCs renewing cycling endometrium is concentrated in the JZ as well. Thus both UL and EM tumors have mSCs as their progenitors which travel out of the myometrium/endometrium JZ of the uterus. Under some unfavorable conditions () these mSCs undergo blastocyst transformation and thus can give rise to either UL or EM.
The precise sequence of molecular events accompanying tumorigenicity in EM or UL though still hypothetical also reveals many features in common. It includes activation of antiapoptotic and mitogenic pathways, changes of inter cell communication, storage of excessive extracellular matrix, increase in cell migration, and angiogenesis. Each of them though rather specific by itself finalize at the same end point. Both disorders contain identical or functionally very close genes and gene families of embryonic development (WNT, HOX), steroid sex hormones and their receptors (PRβ,ESR1,ERβ), epithelium and stromal cell adhesion genes (FN1,VETZ), collagen genes, oncogenes and oncosuppressors (P53, CDKN2b), as well as the moiety of common specific growth factors, apoptotic and antiapoptotic regulators ().
EMT looks as an important prerequisite of initial stages of tumor progression in EM and mutations of MED12 or HMGA genes are the most feasible drivers of UL. Activation of the WNT/β-catenine pathway being important for mytogenic stimulation is also shared by both tumors [Mas et al. Citation2014]. Similar molecular mechanisms underlie expansion of ECM with activation of the metalloproteinase gene family as well as many genes responsible for the growth of ECM.
Epigenetic regulation of gene expression in EM and UL such as DNA methylation or micro-RNAs have many similar features. Global abnormal methylation/ demethylation of the genes, abnormal profiles of regulatory micro-RNA, and dysregulation of immunological reactions are shared by both disorders. In EM and also UL local inflammation induces the expression of TNFα and other cytokines (IL-12, IL-4 as well as their receptors), provokes deregulation of the genes of progesterone receptor, steroid protein of the acute phase, revertase (CYP2C19), and affects oncosuppressive genes CDKN2B and CDKN2A and the genes of early embryonic development HOXA, WNT4. Dysregulation of methylation results in their functional impairment. As already mentioned (see above) ‘syntropy’ means nonrandom combination of two or more genetically related pathological configurations sharing many common molecular mechanisms underlying their origin and progression. Both EM and UL fit well all these demands, as they include: i) identity or obvious similarity of their progenitor stem cells, ii) feasible common site of their origin – JZ; iii) rather similar sequence of molecular events accompanying tumorigenicity, and iv) dysregulation of hormonal and immunological reactions due to global abnormal methylation/ demethylation of the relevant genes, abnormal profiles of regulatory micro-RNA.
Thus there is little doubt that EM and UL should be considered as syntropic diseases. However, it is worth stressing that though rather similar at the start, the pathways of EM and UL progression quickly come apart and lead to quite distinct disorders each with its own unique clinical manifestation. Some of these differences have been presented above.
Epigenetic landscape
At the top of the scheme shown in , one can see a classic image of ‘epigenetic landscape’ initially suggested to explain canalized differentiation of early embryonic cells to its final destination point [Waddington Citation1966]. The type of the channel and the end point of differentiation are under the control of numerous genetic and epigenetic factors, which gradually narrow prospective options of initially omnipotent embryonic cell. Unfavorable genetic or epigenetic factors can drastically change the developmental program and provoke a disease [Baranov et al. Citation2015].
The situation with initial mSC giving rise to EM and UL looks rather similar. Blastocyst transformation and subsequent development of mSC are specified by their former epigenetic history, that is by their position within the JZ of the uterus. Transformed mSC from the inner, endometrial side of the JZ give rise to EM tissue while their counterparts from the outer myometrial side are prone to generate LM ().
After the onset of pathological process induced by some intrinsic or external factors tumor development proceeds to clinically different forms of EM or UL under the constant control of many variable genetic and epigenetic factors. Whether under certain conditions mSCs could give rise not only to different clinical forms of EM and UL but also to adenomyosis (AM), endometrial hyperplasia, malignant tumors or even to ‘endometrial-subendometrial myometrium disruption disease’ remains unknown at present but according to the epigenetic landscape model these option cannot be excluded.
Concluding remarks
Comparative analysis leaves little doubt that EM and UL should be treated as syntropic diseases. Identical or very similar molecular mechanisms underlying both disorders become obvious starting from the origin of their metaplasia and progression to EM and UL lesions. The JZ demarcating endometrial/myometrium boundary in the uterus wall is hypothesized as a source of these mSCs. Irrespective of their precise molecular mechanisms of the origin, subsequent development of mSCs is running under the control of many variable growth factors (hormonal, humoral, immune, and autocrine or paracrine nature) which can substantially depart from the epigenetic landscape of the diseases. Multiple and highly variable clinical forms of EM and UL as well as peculiarities of their unique personal manifestation could be explained by the variability of genetic and epigenetic factors regulating mSCs development.
Stronger associations of GWAS detected EM genes with Stage III/IV emphasizes the importance of detailed sub-phenotype collections of EM samples to allow discrimination of genetic or epigenetic factors associated with different forms of disease [Rahimoglou et al. Citation2014]. Representative bio-banks of EM and UL patients with emphasis on particular phenotypic profiles supplemented with thorough genetic analysis can help to elucidate molecular mechanisms underlying different clinical forms of the disorders and their personal variations. Comparative Next Generation Sequencing (NGS) of paired samples of normal and affected tissues should also be helpful in elucidating molecular mechanisms responsible for EM and UL.
As might be inferred special attention should be paid to the properties of mSC, experimental studies of intrinsic and extrinsic factors facilitating their blastocyst transformation. Special attention should be given to clarify the meaning of mSC’s position within the JZ zone of the uterus as well as to factors stimulating migration of mSC and molecular mechanisms of their metaplasia.
More systems genetics studies are needed to understand how DNA variations in mSC are translated into RNA, protein, and metabolite levels to get more knowledge on common and specific molecular mechanisms underlying pathogenesis and pathophysiology of EM and UL. Transition from genetic to genomic and further to postgenomic technologies such as NGS [Montgomery et al. Citation2014; Miller et al. Citation2015], integrated systems genetics approaches [Baranov et al. Citation2015] and sophisticated signaling pathway analyses [Borahay et al. Citation2015] should result in tremendous progress in our understanding of the genetic architecture of EM and UL and facilitate their early diagnostics, efficient treatment, and prevention.
Declaration of interests
The authors report no conflict of interest. The manuscript preparation was supported by Russian Scientific Foundation (grant number 14-15-00737).
Acknowledgments
The authors acknowledge Dr. Olga Malysheva for fruitful discussions and useful advice and Professor Tatyana Kuznetzova and Dr. Olga Efimova for assistance in preparing the article and for helpful comments.
Additional information
Notes on contributors
Vladislav S. Baranov
Conception and design, revising manuscript for critical comments: VSB; Drafting manuscript: TEI, MIY; References list revision: MIY; Figure drawings: TEI. All authors approved revisions and the final article.
References
- Aznaurova, Y.B., Zhumataev, M.B., Roberts, T.K., Aliper, A.M. and Zhavoronkov, A.A. (2014) Molecular aspects of development and regulation of endometriosis. Reprod Biol Endocrinol 12: 50.
- Baranov, V.S., Ivaschenko, T.E., Liehr, T. and Yarmolinskaya M.I. (2015) System genetics view of endometriosis: a common complex disorder. Eur J Obstet Gynecol Reprod Biol 185: 59–65.
- Beliard, A., Noel, A. and Foidart, J.M. (2004) Reduction of apoptosis and proliferation in endometriosis. Fertil Steril 82: 80–85.
- Bertsch, E., Qiang, W., Zhang, Q., Espona-Fiedler, M., Druschitz, S., Liu, Y., et al. (2014) MED12 and HMGA2 mutations: Two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod Pathol 27: 1144–1153.
- Benassayag, C., Leroy, M. J., Rigourd, V., Robert, B., Honore, J. C., Mignot, T. M., et al. (1999) Estrogen receptors (ERα/ERβ) in normal and pathological growth of the human myometrium: pregnancy and leiomyoma. Am J Physiol Endocrinol Metab 276: E1112–E1118.
- Bischoff, F.Z., Heard, M. and Simpson, J. L. (2002) Somatic DNA alterations in endometriosis: high frequency of chromosome 17 and p53 loss in late-stage endometriosis. J Reprod Immunol 55: 49–64.
- Borahay, M.A., All-Hendy, A.A., Killic, G.S. and Boehning D. (2015) Signalling pathways in leiomyoma: understanding and implication for therapy. Mol Med 21: 242–256.
- Bulun, C. (2013) Uterine Leiomyoma. N Engl J Med 369: 1344–1355.
- Burney, R.O.and Giudice, L.C. (2012) Pathogenesis and pathophysiology of endometriosis. Fertil Steril 98(3): 511–519.
- Canis, M., Donnez, J. G., Guzick, D. S., Halme, J. K., Rock, J. A., Schenken, R. S., et al. (1997) Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil Steril 67(5): 817–821.
- Chan, R.W., Schwab, K.E. and Gargett, C.E. (2004) Clonogenicity of human endometrial epithelial and stromal cells. Biol Reprod 70:1738-1750.
- Chang, J.H., Au, H-K., Lee, W.C., Chi, C.C., Ling, T.Y, Wang, L.M., et al. (2013) Expression of the pluripotent transcription factor OCT4 promotes cell migration in endometriosis. Fertil Steril 99: 1332–1339.
- Ciavattini, A., Giuseppe, J.D., Storton, P., Montik, N., Giannubilo, S. R., Litta, P., et al. (2013) Uterine fibroids: pathogenesis and interactions with endometrium and endomyometrial junction. Obstet Gynecol Int 2013: 173184.
- Dyson, M.T., Roqueiro, D. and Monsivais, D. (2014) Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet 10: e1004158.
- Forte, A., Cipollaro, M. and Galderisi, U. (2014) Genetic, epigenetic and stem cell alterations in endometriosis: new insights and potential therapeutic perspectives. Clin Sci (Lond) 126: 123–138.
- Fujimota, J., Hirose, R., Ichigo, S., Sakaguchi, H., Li, Y. and Tamaya, T. (1998) Expression of progesterone receptor form A and B mRNAs in uterine leiomyoma. Tumor Biol 19: 126–131.
- Fung, J.N., Holdsworth-Carson, S.J., Sapkota, Y., Zhao, Z.Z., Jones, L., Girling, J.E., et al. (2015) Functional evaluation of genetic variants associated with endometriosis near GREB1. Hum Reprod 30(5): 1263–1275.
- Gargett, C.E., Schwab, K.E., Zillwood, R.M., Nguyen, H.P. and Wu, D. (2009) Isolation and culture of epithelial progenitors and mesenchymal stem cells from human endometrium. Biol Reprod 80: 1136–1145.
- Gotte, M., Wolf, M., Staebler, A., Buchweitz, O., Kelsch, R., Schuring A.N., et al. (2008) Increased expression of the adult stem cell marker Musashi-1 in endometriosis and endometrial carcinoma. J Pathol 215: 317–329.
- Grings, A.O., Lora, V., Ferreira, G.D., Brum, I.S., Corleta, H. and Capp, E. (2012) Protein expression of estrogen receptors α and β and aromatase in myometrium and uterine leiomyoma. Gynecol Obstet Invest 73: 113–117.
- Holzmann, C., Markowski, D.N., Koczan, D., Küpker, W., Helmkeand, B.M. and Bullerdiek, J. (2014) Cytogenetically normal uterine leiomyomas without MED12-mutations – a source to identify unknown mechanisms of the development of uterine smooth muscle tumors. Mol Cytogenet 7: 88–96.
- Houshdaran, S., Zelenko, Z., Irwin, J.C. and Giudice, L.C. (2014) Human endometrial DNA methylome is cycle- dependent and is associated with gene expression regulation. Mol Endocrinol 28: 1118–1135.
- Huang, H.P., Chen, P.H., Yu, C.Y., Chuang, C.Y., Stone, L., Hsiao, W.C., et al. (2011) Epithelial cell adhesion molecule (EpCAM) complex proteins promote transcription factor-mediated pluripotency reprogramming. J Biol Chem 286: 33520–33532.
- Jolinière, J.B., Ayoubi, J.M.B., Gianaroli, L., Dubuisson, J.B., Gogusev, J. and Feki, A. (2014) Endometriosis: A new cellular and molecular genetic approach for understanding the pathogenesis and evolutivity. Front Surg 1: 16.
- Kalluri, R. (2009) EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest 119: 1417–1419.
- Kim, J.J. and Sefton, E.C. (2012) The role of progesterone signaling in the pathogenesis of uterine leiomyoma. Mol Cell Endocrinol 358: 223–231.
- Kim, S., Xu, X., Hecht, A. and Boyer, T.G. (2006) Mediator is a transducer of Wnt/beta-catenin signaling J Biol Chem 281: 14066–14075.
- Kobayashi, H. (2014) Imprinting genes associated with endometriosis. EXCLI J 13:252-264.
- Kobayashi, H, Iwai, K., Niiro, E., Morioka, S. and Yamada, Y. (2014) Fetal programming theory: implication for the understanding of endometriosis. Hum Immunol 75: 208–217.
- Kocbek, V., Vouk, K., Bersinger, N.A., Mueller, M.D. and Rizner, L.T. (2015) Panels of cytokines and other secretory proteins as potential biomarkers of ovarian endometriosis. J Mol Diagn 17: 325–334.
- Kunz, G., Beil, D., Huppert, P. Noe, M., Kissler, S. and Leyendecker, G. (2005) Adenomyosis in endometriosis – prevalence and impact on fertility. Evidence from magnetic resonance imaging. Human Reprod 20:2309- 2316.
- Larsen, S.B., Lundorf, E., Forman, A. and Dueholm, M. (2011) Adenomyosis and junctional zone changes in patients with endometriosis. Eur J Obstet Gynecol Reprod Biol 157: 206–211.
- Laughlin, S.K., Herring, A.H., Savitz, D.A., Olshan, A.F., Fielding, J.R., Hartmann, K.E., et al. (2010) Pregnancy-related fibroid reduction. Fertil Steril 94: 2421–2423.
- Leppert, P.C., Catherino, W.H. and Segars, J.H. (2006) A new hypothesis about the origin of uterine fibroids based on gene expression profiling with microarrays. Am J Obstet Gynecol 195: 415–420.
- Mäkinen, N., Vahteristo, P., Bützow, R., Sjöberg, J. and Aaltonen, L.A. (2013) Exomic landscape of MED12 mutation-negative and –positive uterine leiomyomas. Intern J Cancer 134: 1008–1012.
- Malik, M. and Catherino, W.H. (2012) Development and validation of a three-dimensional in vitro model for uterine leiomyoma and patient-matched myometrium. Fertil Steril 97:1287–1293.
- Malik, M., Norian, J., McCarthy-Keith, D., Britten, J. and Catherino, W.H. (2010) Why leiomyomas are called fibroids: the central role of extracellular matrix in symptomatic women. Semin Reprod Med 28: 169–179.
- Marsh, E.E., Lin, Z., Yin, P., Milad, M., Chakravarti, D. and Bulun, S.E. (2008) Differential expression of microRNA species in human uterine leiomyoma versus normal myometrium. Fertil Steril 89: 1771–1776.
- Markowski, D.N., Helmke, B.M., Belge, G., Nimzyk, R., Bartnitzke, S., Deichert, U., et al. (2011) HMGA2 and p14Arf: major roles in cellular senescence of fibroids and therapeutic implications. Anticancer Res 31: 753–761.
- Markowski, D.N., Helmke, B.M., Bernitzke, S., Loning, T. and Bullerdierk J. (2014) Uterine fibroids – do we deal with more than one disease? Int J Gynecol Pathol 33(6): 568–572.
- Maruyama, T. (2014) Endometrial stem/progenitor cells. J Obstet Gynec Res 40: 2015–2022.
- Mas A., Cervello I., Gill-Sanchis, C. and Sim, C. (2014) Current understanding of somatic stem cells in leiomyoma formation. Fertil Steril 102(3):613–620.
- Matsuzaki, S. and Darcha, C. (2012) Epithelial to mesenchymal transition-like and mesenchymal to epithelial transition-like processes might be involved in the pathogenesis of pelvic endometriosis. Hum Reprod 27: 712–721.
- Miller, N.A., Farrow, E.G., Gibson, M., Willig, L.K., Twist, G., Yoo, B., et al. (2015) A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic disease. Genome Med 7: 100.
- Montgomery, G.W., Zondervan, K.T. and Nyholt, D.R. (2014) The future for genetic studies in reproduction. Mol Hum Reprod 20: 1–14.
- Moroni, R.M., Vieira, C.S., Ferriani, R.A., Reis, R.M., Nogueira, A.A. and Brito, L.G. (2015) Presentation and treatment of uterine leiomyoma in adolescence: a systematic review. BMC Womens Health 15: 4.
- Norian, J.M., Malik, M., Parker, C.Y., Joseph, D., Leppert, P.C. and Segars, J.H. (2009) Transforming growth factor beta3 regulates the versican variants in the extracellular matrix-rich uterine leiomyomas. Reprod Sci 16: 1153–1164.
- Norian, J.M., Owen, C.M. and Taboas, J. (2012) Characterization of tissue biomechanics and mechanical signaling in uterine leiomyoma. Matrix Biol 31: 57–65.
- Nothnick, W.B., Al-Hendy, A. and Lue, J.R. (2015) Circulating microRNAs as diagnostic biomarkers for endometriosis: privation and promise. J Minim Invasive Gynecol 22(5): 719–726.
- Okamoto, M., Nasu1, K., Abe, W., Aoyagi, Y., Kawano, Y., Kai, K., et al. (2014) Enhanced miR-210 expression promotes the pathogenesis of endometriosis through activation of signal transducer and activator of transcription 3. Hum Reprod 30(3):632–641.
- Okolo, S. (2008) Incidence, aetiology and epidemiology of uterine fibroids. Best Pract Res Clin Obstet Gynaecol 22(4): 571–588.
- Ono, M., Qiang, W., Serna, V.A., Yin, P., Coon, J.S., Navarro, V.A., et al. (2012) Role of stem cells in human uterine leiomyoma growth. PLoS ONE 7(5): e36935.
- Osinovskaya, N.S., Malysheva, O.V., Shved, N.Yu., Ivashchenko, T.E., Sultanov, I.Yu., Efimova, O.A., et al. (2015) Frequency and spectrum of MED12 exon 2 mutations in multiple versus solitary uterine leiomyomas from Russian patients. Int J Gynecol Pathol ( in press).
- Padykula, H.A. (1991) Regeneration in the primate uterus: the role of stem cells. Ann N Y Acad Sci 622: 47–56.
- Pellegrini, C., Gori, I., Achtari, C., Hornung, D., Chardonnens, E., Wunder, D., et al. (2012) The expression of estrogen receptors as well as GREB1, c-MYC, and cyclin D1, estrogenregulated genes implicated in proliferation, is increased in peritoneal endometriosis. Fertil Steril 98: 1200–1208.
- Pfaundler, M. and von Sehr, L. (1922) Weiteres uber Syntropie kindlicher Krankheitzzustande. Z Kinderheilkd 30: 298–313.
- Pittatore, G., Moggio, A., Benedetto, C., Bussolati, B. and Revelli, A. (2013) Endometrial Adult/Progenitor Stem Cells: Pathogenetic Theory and New Antiangiogenic Approach for Endometriosis Therapy. Reprod Sci 21(3):296–304.
- Proestling, K., Birner, P., Gamperl, S., Nirtl, N, Marton, E., Yerlikaya, G., et al. (2015) Enhanced epithelial to mesenchymal transition (EMT) and upregulated MYC in ectopic lesions contribute independently to endometriosis. Reprod Biol Endocrinol 13: 75.
- Puzyrev, V.P., Makeeva, O.A. and Freidin, M.B. (2010) Syntropy, genetic testing and personalized medicine. Personalized Med 7: 399–405.
- Rahmioglu, N., Nyholt, D.R., Morris, A.P., Missmer, S.A., Montgomery, G.W. and Zodervan, K.T. (2014) Genetic variants underlying risk of endometriosis: insight from meta-analysis of eight genome-wide association and replication datasets. Hum Reprod Update 20: 702–716.
- Sasson, I.E. and Taylor, H.S. (2008) Stem cells and the pathogenesis of endometriosis. Ann NY Acad Sci 1127: 106–115.
- Schwab, K.E., Hutchinson, P. and Gargett, C.E. (2008) Identification of surface markers for prospective isolation of human endometrial stromal colony-forming cells. Hum Reprod 23: 934–943.
- Signorilea, P.G. and Baldia, A. (2015) New evidence in endometriosis. Int J Biochem Cell Biol 60: 19–22.
- Sotomayor, P., Godoy, A., Smith, G.J. and Huss, W.J. (2009) Oct4A is expressed by a subpopulation of prostate neuroendocrine cells. Prostate 69:401–410.
- Su, J., You, P., Li, W.L., Tao, X.R., Zhu, H.Y., Yao, Y.C., et al. (2010) The existence of multipotent stem cells with epithelial-mesenchymal transition features in the human liver bud. Int J Biochem Cell Biol 42: 2047–2055.
- Sun, F.Q., Duan, H., Wang, S. and Li, J.J. (2015) 17β-Estradiol Induces Overproliferation in Adenomyotic Human Uterine Smooth Muscle Cells of the Junctional Zone Through Hyperactivation of the Estrogen Receptor-Enhanced RhoA/ROCK Signaling Pathway. Reprod Sci 22(11): 1436–1444.
- Suryawanshi, S., Vlad, A.M., Lin, H.M. and Mantia-Smaldone, G. (2013) Plasma microRNAs as novel biomarkers for endometriosis and endometriosis-associated ovarian cancer. Clin Cancer Res 19: 1213–1224.
- Tal, R. and Segars, J.H. (2014) The role of angiogenic factors in fibroid pathogenesis: potential implications for future therapy. Hum Reprod Update 20: 194–216.
- Thiery, J.P., Acloque, H., Huang, R.Y. and Nieto, M.A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139: 871–890.
- Tocci, A., Greco, E. and Ubaldi, F.M. (2008) Adenomyosis and endometrial-ubendometrial myometrium unit disruption disease are two different entities. Reprod Biomed Online 17: 285–291.
- Tulac, S., Nayak, N.R., Kao, L.C., VanWaes, M., Huang, J., Lobo, S., et al. (2003) Identification, characterization, and regulation of the canonical Wnt signaling pathway in human endometrium. J Clin Endocrinol Metab 88: 3860–3866.
- Waddington, C.H. (1966) Principles of development and differentiation. New York, Macmillan Pub Co Inc.
- Wang, T., Zhang, X., Obijuru, L., Laser, J., Aris, V., Lee, P., et al. (2007) A micro-RNA signature associated with race, tumor size, and target gene activity in human uterine leiomyomas. Genes Chromosomes Cancer 46: 336–347.
- Wei, J., Chiriboga, L. and Mittal K. (2005) Expression profile of the tumorigenic factors associated with tumor size and sex steroid hormone status in uterine leiomyomata. Fertil Steril 84: 474–484.
- Yamagata, Y., Maekawa, R. and Asada, H. (2009) Aberrant DNA methylation status in human uterine leiomyoma. Mol Hum Reprod 15: 259–267.
- Yang, J., Mani, S.A., Donaher, J.L., Ramaswamy, S., Itzykson, R.A. and Come, C. (2004) Twist, a master regulator 380 of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939.
- Yang, W., Zhang, Y., Fu, F. and Li, R. (2013) High-resolution array-comparative genomic hybridization profiling reveals 20q13.33 alterations associated with ovarian endometriosis. Gynecol Endocrinol 29: 603–607.
- Ying, Z. and Weiyuan, Z. (2009) Dual actions of progesterone on uterine leiomyoma correlate with the ratio of progesterone receptor A:B. Gynec Endocrinol 25: 520–523.
- Zhang, Y., Yu, P., Sun, F., Li, T.C., Cheng, Jm. and Duan H. (2015) Expression of oxytocin receptors in the uterine junctional zone in women with adenomyosis. Acta Obstet Gynecol Scand 94(4): 412–418.
- Zondervan, K.T., Cardon, L.R. and Kennedy, S.H. (2001) The genetic basis of endometriosis. Curr Opin Obstet Gynecol 13: 309–314.
- Zondervan, K.T, Cardon, L.R. and Kennedy, S.H. (2002) What makes a good case-control study? Design issues for complex traits such as endometriosis. Hum Reprod 17: 1415–1423.