Abstract
DNA barcoding is a method promising fast and accurate identification of animal species based on the sequencing of the mitochondrial c oxidase subunit (COI) gene. In this study, we explore the prospects for DNA barcoding in one particular fish group, the billfishes (suborder Xiphioidei—swordfish, marlins, spearfishes, and sailfish). We sequenced the mitochondrial COI gene from 296 individuals from the 10 currently recognized species of billfishes, and combined these data with a further 57 sequences from previously published projects. We also sequenced the rhodopsin gene from a subset of 72 individuals to allow comparison of mitochondrial results against a nuclear marker. Five of the 10 species are readily distinguishable by COI barcodes. Of the rest, the striped marlin (Kajikia audax) and white marlin (K. albida) show highly similar sequences and are not unambiguously distinguishable by barcodes alone, likewise are the three spearfishes Tetrapturus angustirostris, T. belone, and T. pfluegeri. We discuss the taxonomic status of these species groups in light of our and other data, molecular and morphological.
Introduction
In the continuing quest to catalog and monitor the biodiversity of the world's oceans, molecular methods are assuming an increasing prominence. Fishes constitute the largest vertebrate group on the planet, with approximately 30,000 species (Nelson Citation2006), and fisheries and related industries are vital to the economies of many nations providing a significant fraction of global human food needs. Accurate and reliable species identifications of fish and fish tissues are important in many situations, including the detection of market substitutions (in which fish is sold under an incorrect species name, either to increase profit or to conceal illegal catches) (Wong and Hanner Citation2008; CitationGAO 2009), monitoring of fisheries landings for stock assessments and management, early life stage assessments (Victor et al. Citation2009), and scientific research generally (Yancy et al. Citation2008). Fish species identification has traditionally been carried out based on the examination of specimen morphology; however, in many cases in which species identifications are needed, morphological features may not be available (e.g. when food inspectors wish to determine whether a piece of fillet sold at a market is correctly named), or not yet developed (e.g. in which fish eggs or larvae need to be assigned to species). In such cases in which morphological information is lacking, molecules (DNA, RNA, and proteins) may serve as unique identifiers to discriminate among species (Arnot et al. Citation1993; Floyd et al. Citation2002).
In the past, a variety of molecular techniques have been utilized to properly discriminate among species, including protein electrophoresis, nucleic acid ‘fingerprinting’ methods such as restriction fragment length polymorphism (RFLP) and Amplification fragment length polymorphism (AFLP), and direct sequencing of specific genomic regions (Hsieh et al. Citation2005, Citation2007). These methods have generally been of small scale, developed on a case-by-case basis by different research groups and applicable only to small groups of species. In contrast, DNA barcoding is a sequencing-based approach that attempts to establish a single global system of both laboratory protocols and data management, which should ultimately be applicable to all animals (Hebert et al. Citation2003a).
In animals, DNA barcoding is based on sequencing a ∼658 base-pair fragment of the mitochondrial gene, cytochrome c oxidase subunit 1 (COI; Hebert et al. Citation2003a,Citationb). This signature sequence can be compared against a database of known sequences from reference specimens to obtain a species identification (Ekrem et al. Citation2007). Because this method depends on molecular approaches rather than on morphology, it may be applied to organisms of any life stage from egg to adult. It also aims to employ standardized protocols that may be applied to a wide range of organisms by individuals possessing a minimum amount of technical expertise and without the need for extensive knowledge of traditional morphological taxonomy. Sequence and specimen data are stored and made available in the Barcode of Life Data System (BOLD) database (Ratnasingham and Hebert Citation2007), an online collaborative workbench used to construct and curate a global barcode reference sequence library. Under the organizing framework of the International Barcode of Life (iBOL) project, global campaigns are under way to barcode the world's biodiversity, including Fish Barcode of Life (FISH-BOL), the campaign to barcode fishes (Costa and Carvalho Citation2007; Ward et al. Citation2009). Here, we discuss the prospects for DNA barcoding in one particular fish group, the billfishes (suborder Xiphioidei).
The billfishes are a group of ray-finned fishes (class Actinopterygii) comprising two extant families: the monotypic Xiphiidae (swordfish, Xiphias gladius) and Istiophoridae (marlins, spearfishes, and sailfish) (Collette et al. Citation2006). All are large, active apex predators characterized by an elongate, sword-like, or spear-like snout. All species are dioecious (having separate sexes) and females attain larger sizes than males, but otherwise do not display strong sexual dimorphism (CitationCollette, 2010). They are among the largest and fastest swimming bony fishes, and are capable of trans-oceanic movements (Sedberry and Loefer Citation2001). Billfishes are wide ranging, inhabiting both tropical and temperate waters, and, seasonally, also cold waters (to which they migrate during summer months for feeding); spawning of most species occurs in tropical and subtropical waters (CitationNakamura 1985).
All billfish species are of commercial value with particular importance in some Asian markets (Hsieh et al. Citation2005; Gentner Citation2007). Furthermore, all species are also highly desirable targets in recreational fisheries; just in the USA, for example, expenditures by recreational billfish anglers are estimated to exceed $2 billion annually (SEFSC/NMFS Citation2004). Commercial long-line fisheries cover nearly the entire natural distributions of the species, whereas sport fishing grounds are more restricted. Areas where the two zones overlap have been subject to conflicts over fishing rights (CitationNakamura 1985). Populations of all billfish species investigated have declined in recent years due to high levels of human exploitation, and international fishery management organizations have highlighted the need for more fisheries and basic biological information, including better tracking of landings by species and identification of spawning habitats, to aid global billfish management and conservation (Gentner Citation2007; Mora et al. Citation2009).
Although billfishes are both targeted in specific international fisheries and caught extensively as bycatch in pelagic fisheries, few countries keep accurate track of their landings, and even when they do, often group species together (e.g. sailfish and spearfishes) in their landings records (SEFSC/NMFS Citation2004; Gentner Citation2007). Monitoring landings by species even for adult fish is further complicated by the difficulties in identifying processed fishes that end up as similar looking carcasses or smaller body parts in the trade pipeline (McDowell and Graves Citation2002; SEFSC/NMFS Citation2004; Hsieh et al. Citation2005). Delineation of billfish spawning habitat is typically accomplished by conducting oceanographic surveys to find and monitor the temporal and spatial distributions of individuals across life-stages, including eggs, larvae, and adults (Kawakami et al. Citation2010; Richardson et al. Citation2010). Yet identification of billfish early life stages (eggs and larvae) based on morphological characters is often problematic (SEFSC/NMFS Citation2004; Hyde et al. Citation2005).
These species identification difficulties in the context of the ecological and fishery importance of billfishes have led to the development of a variety of genetic approaches to facilitate the accurate identification of their body parts and early life stages. Approaches developed to date include restriction fragment length polymorphism analysis of polymerase chain reaction (PCR) amplified loci, multiplex PCR, and direct sequencing of either the mitochondrial 16S rRNA or cytochrome b loci (McDowell and Graves Citation2002; Hsieh et al. Citation2005; Hyde et al. Citation2005; Luthy et al. Citation2005; Richardson et al. Citation2007; Kawakami et al. Citation2010). These approaches successfully discriminate among the subset of billfish species tested in each study; however, none of these approaches have been applied to all known billfish species. In particular, the white marlin and striped marlin (Kajikia albida/K. audax), which are believed (based on both morphological and molecular comparisons) to have diverged very recently in evolutionary time (Collette et al. Citation2006), have proven problematic for molecular-based discrimination. Previous studies (e.g. Graves Citation1998) have failed to resolve this species pair utilizing genetic sequence data derived from multiple loci, including the mitochondrial genes ND4 and cytochrome b (Shivji et al. Citation2006), and the mitochondrial control region (mtCR) and ND2 and 12S rRNA genes (Collette et al. Citation2006).
Given the goal of establishing a global, standard genetic marker and method for all fishes under the organizing framework of the iBOL initiative, we aim to calibrate the utility of DNA barcoding (using the COI marker) in this important group, evaluating it against a nuclear marker (Rhodopsin) that has also been recommended for large-scale use (Sevilla et al. Citation2007).
Materials and methods
Samples
Specimens were obtained from a variety of sources. Because of the challenges associated with the retention of large-bodied animals and due to the fact that many of the samples derived from fisheries and sport fishing initiatives, there are no morphological voucher specimens associated with these samples. Species identifications were made on the basis of morphology by the collectors at the time of collection. All fish tissue voucher samples (muscle or fin clips) are deposited in the Guy Harvey Research Institute (Dania Beach, FL, USA).
DNA extraction, PCR amplification, and sequencing: Mitochondrial COI gene
DNA was extracted from these samples using an automated glass fiber protocol (Ivanova et al. Citation2006). The DNA barcode region of COI (652 bp) was then amplified by PCR using M13-tailed fish primer cocktails (Ivanova et al. Citation2007). Each 12.5 μl PCR comprised 6.25 μl of 10% trehalose; 2 μl of ultrapure water; 1.25 μl of Invitrogen 10 × PCR buffer [200 mM Tris–HCl (pH 8.4), 500 mM KCl]; 0.625 μl MgCl2 (50 mM); 0.125 μl of each primer cocktail (C_FishF1t1 and C_FishR1t1 from Ivanova et al. Citation2007); 0.062 μl of each dNTP (10 mM); 0.060 μl of Platinum Taq DNA Polymerase (Invitrogen, USA); and 2.0 μl of extracted fish DNA template. Thermocycling conditions were as follows: 95°C for 2 min; 35 cycles [94°C for 30 s, 52°C for 30 s, 72°C for 1 min]; and 72°C for 10 min (hold at 4°C).
PCR products were visualized on pre-cast 1.2% Agarose E-gels (Invitrogen) to check results, and successful amplicons were sequenced bidirectionally using the primers M13F and M13R (Ivanova et al. Citation2007) with BigDye 3.1 Cycle Sequencing mix (Applied Biosystems, USA), and run on an ABI 3730 capillary sequencer to generate electropherogram trace files. Sequences were quality checked, edited, and assembled using SeqScape software (Applied Biosystems), before being uploaded to the Barcode of Life Data Systems (Ratnasingham and Hebert Citation2007). All sequences, as well as trace files and associated specimen data, are available in the project ‘Billfish and Swordfish COI identification’ (EBFSF) at http://www.boldsystems.org; sequences are also deposited in GenBank with the accession numbers (pending).
Previously published COI barcode sequences
Using the BOLD's Taxonomy Search function, all COI sequences in public projects deriving from the members of the families Xiphiidae and Istiophoridae were extracted and added to our analysis. Data were drawn from the following projects on BOLD: Fishes of Australia Part I (FOA); Marine Fishes of California (MFC); Marine Fish of Mexico II (MXII); DNA Barcoding the Indian Marine Fishes (WLIND); GenBank Fish (ANGBF); and Overlooked Fishes in Marine Settings (TZSAA).
DNA extraction, PCR amplification, and sequencing: Nuclear rhodopsin gene
Genomic DNA was extracted from all samples using DNeasy Kits (QIAGEN, Inc., Valencia, USA) from approximately 25 mg of tissue. Amplification via PCR targeted a 460 bp segment of the rhodopsin gene (Rho), suggested by Sevilla et al. (Citation2007) as a potential nuclear barcode for teleost fishes. The primer pair Rod-F2w (5′-AGCAACTTCCGCTTCGGTGAGAA-3′) and Rod-R4n (5′-GGAACTGCTTGTTCATGCAGATGTAGAT) (Sevilla et al. Citation2007) were utilized to both amplify and sequence the DNA segment. Total PCR volumes were 50 μl and contained 33.3 μl of high purity water (OmniSolv®, VWR, Radnor, USA), 5 μl 10 × PCR buffer [15 mM MgCl2], 8 μl dNTPs [1.25 mM of each dNTP], 1.25 μl of each primer (10 pmol), 1 unit of HotStar Taq DNA Polymerase (QIAGEN, Inc.), and 1 μl of template genomic DNA. PCR was carried out in a Mastercycler Gradient (Eppendorf, Inc., New York, USA) thermal cycler as follows: 95°C initial heating for 15 min; 35 cycles [94°C for 1 min, 50°C for 1 min, and 72°C for 2 min]; 72°C for 20 min.
A negative control (no genomic DNA) was included in each set of reactions to check for reagent contamination. PCR products were visualized on a 1.2% agarose gel to check for successful amplification, and were subsequently purified using the QIAquick PCR Purification Kit (QIAGEN, Inc.). Sequencing was carried out using BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), and all samples were run on an automated ABI 3130 genetic analyzer. Sequences were aligned manually using the program Genedoc 2.6.002 (CitationNicholas and Nicholas 1997), and were subsequently uploaded to the BOLD database. As indicated above, all sequences (GenBank Accession nos. pending), trace files, and specimen data are available in BOLD.
Phylogenetic reconstruction
The program MEGA 4.0.2 (Kumar et al. Citation2008) was used to generate a neighbor-joining (NJ) phenogram using the Kimura 2-parameter (K2P) distance metric to visually compare sequence similarities. To examine statistical support for branches, we reduced the dataset to a single representative for each unique haplotype (47 sequences), and a maximum parsimony analysis was carried out in MEGA, including bootstrap resampling with 100 replicates.
Sequence groups that were not clearly separated on the NJ tree were analyzed using haplotype parsimony networks, generated using the program TCS (Clement et al. Citation2000) at the 99% connection limit.
Reanalysis of previous data
mtCR sequences from two previously published studies (Graves and McDowell Citation2006; McDowell and Graves Citation2008) were downloaded from GenBank. These comprised 79 sequences from striped marlin (K. audax) (Accession Nos DQ199950–DQ200028) and 91 from white marlin (K. albida) (Accession Nos DQ835191–DQ835281). These previous papers only examined genetic variation within these two species separately, and did not combine all sequences into a single analysis.
Results
DNA barcodes (mitochondrial COI)
COI sequences were obtained from 296 individuals from the 10 currently recognized species of billfish: black marlin (Istiompax indica)—12 specimens; blue marlin (Makaira nigricans)—49 specimens; sailfish (Istiophorus platypterus)—34 specimens; white marlin (K. albida)—44 specimens; striped marlin (K. audax)—28 specimens; shortbill spearfish (Tetrapturus angustirostris)—11 specimens; Mediterranean spearfish (T. belone)—15 specimens; roundscale spearfish (T. georgii)—51 specimens; longbill spearfish (T. pfluegeri)—43 specimens; swordfish (X. gladius)—12 specimens. Mean sequence length was 647 bp (range: 500–652 bp). No evidence of indels or stop codons was seen (which if present would suggest amplification of a NUMT rather than the functional mitochondrial COI). Overall, nucleotide frequencies in this dataset were G = 18.82%; C = 29.51%; A = 22.71%; and T = 28.95%.
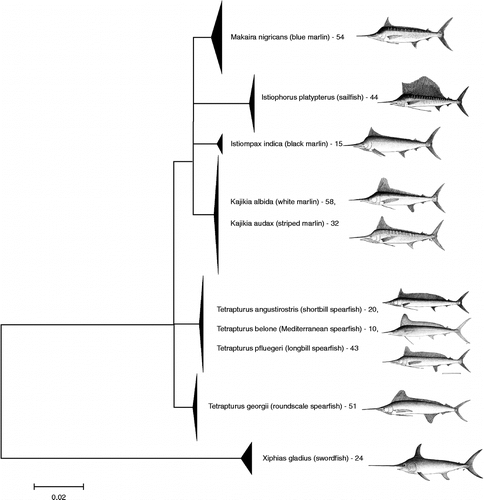
In addition, a further 57 billfish COI sequences from the publically accessible section of BOLD (from previously published barcoding projects or gathered from GenBank) were included in our analysis. A NJ tree of all 353 sequences (K2P distance) showed that 5 of the 10 species form distinct, cohesive clusters (). In contrast, the white and striped marlin pair (K. albida and K. audax) appear mixed in the same cluster, and the same is true for three of the four known spearfishes: T. angustirostris, T. belone, and T. pfluegeri (in contrast, T. georgii was well separated from its congenerics). The maximum parsimony analysis (included in supplementary online material) shows low bootstrap support for the deeper nodes of the tree; therefore, these internal branches were reduced to polytomies on the main NJ tree in . It should be noted that this tree is not intended as a hypothesis of phylogenetic relationships but simply as a visual representation of haplotype groupings.
Figure 1. NJ tree of COI sequences. NJ analysis, carried out in MEGA using K2P distance, of all billfish COI barcode sequences included this study. The full tree of 353 sequences is included in Supplementary Online Material. Multiple sequences are collapsed to triangles in which vertical distance corresponds to the number of sequences, and horizontal distance is proportional to sequence diversity. Numbers indicate the number of individual specimens in a species. Several internal branches were collapsed to polytomies to indicate low bootstrap support for the same nodes in a maximum parsimony analysis—see supplementary online material. Images are taken from the FAO ‘Billfishes of the World’ catalog (CitationNakamura 1985) and are used with the permission of the copyright holder.
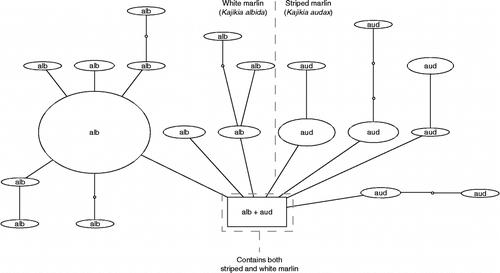
To examine sequence variation more closely within each of these two multi-species groups (white/striped marlins and spearfishes), we constructed haplotype parsimony networks using TCS. For the white and striped marlin (), the 91 sequences formed 21 distinct haplotypes; whereas sequences between the two species were often highly similar (differing by only 1 bp in some cases), only one exact haplotype was shared between K. albida and K. audax; this was also determined in TCS to be the likely root (ancestral) haplotype. The individuals carrying this haplotype included six specimens of K. albida and five of K. audax (). The between-group mean K2P distance between K. albida and K. audax for all COI sequences was 0.004.
Figure 2. Haplotype parsimony network for white and striped marlin. Haplotype parsimony network, generated using the program TCS at the 99% connection limit, of 91 COI sequences belonging to the white marlin (K. albida) and striped marlin (K. audax). Individuals carrying the shared haplotype (square box) included six specimens of K. albida (EBFSF021-07, EBFSF025-07, EBFSF103-09, EBFSF115-09, MXII141-07, MXII138-07) and five of K. audax (EBFSF278-10, EBFSF280-10, EBFSF284-10, EBFSF289-10, EBFSF291-10).
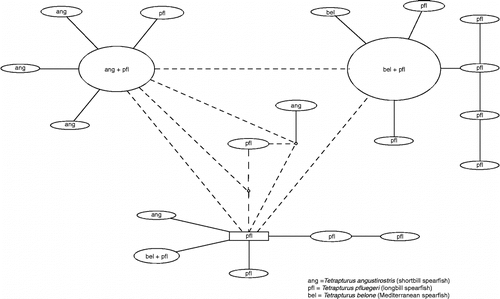
For the spearfishes (), the 64 sequences fell into 16 haplotypes, of which just three were shared by two of the three species (none were shared by all three species). The remaining haplotypes were private to a single species.
Figure 3. Haplotype parsimony network for spearfishes. Haplotype parsimony network, generated using the program TCS at the 99% connection limit, of 64 COI sequences belonging to the spearfish species T. angustirostris, T. belone, and T. pfluegeri.
Reanalysis of mtCR sequences
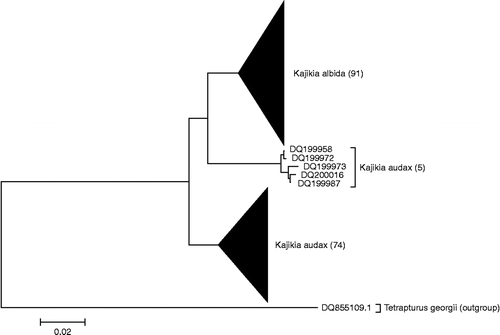
A new NJ tree () of 170 control region (mtCR) sequences from striped marlin (K. audax) and white marlin (K. albida) (Graves and McDowell Citation2006; McDowell and Graves Citation2008) showed that, although most of the sequences clustered into two separate clades by species, a small group (n = 5) of striped marlin sequences shows greater similarity to the white marlin clade than to the rest of the striped marlin sequences.
Figure 4. NJ tree of mtCR sequences. NJ analysis, carried out in MEGA using K2P distance, of mtCR sequences, extracted from GenBank and originally from the studies by Graves and McDowell (Citation2006) and McDowell and Graves (Citation2008).
Nuclear rhodopsin
Sequences of the nuclear Rho were obtained from 72 specimens (all but two also represented by a COI sequence). All sequences were 460 bp in length. Overall nucleotide frequencies in this dataset were G = 25.4%; C = 29.48%; A = 17.82%; and T = 27.22%.
An NJ tree of these 72 sequences () showed that rhodopsin also failed to resolve the albida-audax and the angustirostris-belone-pfluegerii species clusters, and that furthermore, T. georgii is not cleanly separated from the other spearfishes. Although a single fixed nucleotide difference discriminated all nine K. albida specimens from the majority of the K. audax (position 199/460; ‘A’ in albida, ‘G’ in audax; a synonymous substitution), a small number—four individuals—of K. audax appear to be heterozygous at this position (represented ‘R’—the IUPAC code for A or G—in our alignment)—determined by two overlapping peaks at this position in the sequence electropherogram. These specimens were EBFSF035-07, EBFSF129-09, EBFSF130-09, and EBFSF137-09 (). Notably, however, these do not include the same individuals that share the identical COI haplotype, as seen in the TCS analysis (see above).
Figure 5. NJ tree of rhodopsin sequences. NJ analysis of 72 nuclear rhodopsin (Rho) sequences, carried out using the BOLD Taxon ID Tree function, using K2P distance. Arrow indicates the four apparently heterozygous K. audax individuals.

Discussion
These results suggest that at least 5 of the 10 recognized species of billfish are readily distinguishable by standard COI barcodes: I. indica, I. platypterus, M. nigricans, T. georgii, and X. gladius. The remaining five species fall into two complexes within which species may not be unambiguously resolved by barcode sequences: the striped marlin (K. audax) and white marlin (K. albida) are one complex, whereas the other consists of the three spearfishes T. angustirostris, T. belone, and T. pfluegeri. Nevertheless, the presence of private haplotypes () still offers the potential for assigning an unknown specimen to species (though given the relatively small sample size examined thus far, the possibility that more shared haplotypes might be seen with more sequences cannot be ruled out). It is interesting to note that, for the white and striped marlin, the single COI haplotype that is shared by the members of both species was also determined by TCS to be the most likely ancestral haplotype for the whole group. Sampling locations in which this haplotype was recovered included both the Pacific and Atlantic coastal areas of Mexico. Interestingly, when examining the same individuals for both mitochondrial COI and nuclear rhodopsin sequences, it is not the same groups of individuals carrying the identical sequences—those which are indistinguishable by COI are distinguishable by Rho, and vice versa (in which both loci are available from the same individuals).
Our analysis of COI and rhodopsin further underscores the difficulty associated with resolving this species-complex, failing to detect a clear genetic distinction between the two species. Thus, far, all genetic analyses conducted have failed to clearly distinguish the two species as reciprocally monophyletic groups, regardless of the locus examined (Collette et al. Citation2006; this study). Although some genetic differentiation exists among most examined specimens of white and striped marlin, this differentiation is thus far small (mean between-group K2P distance of 0.004 for COI sequences), raising the issue of whether this is sufficient to diagnose the two as separate species. Alternatively, the detection of such limited genetic differentiation may in fact suggest that these two ‘species’ actually comprise two geographically separated subspecies with gene flow still occurring at points of contact, or indeed might simply be different populations of the same species. Indeed, morphologically the white marlin and striped marlin also show a high degree of similarity. In his key, CitationNakamura (1985) separates audax from albidus and georgii just based on the tips of the fins being pointed in audax versus rounded in the other two species. The existence of four genetically discrete populations of striped marlin within the Pacific (McDowell and Graves Citation2008) further complicates the issue of species delineation, as some of these populations may also merit recognition as subspecies. On the basis of the available genetic and morphological evidence, the whole question of the validity of continued recognition of K. albida and K. audax as distinct species, as opposed to subspecies or even populations, requires reexamination with additional morphological and/or molecular data.
Given the strong morphological similarity of the billfishes, the ability to unambiguously resolve species is impeded by a lack of vouchers or associated metadata (especially for sequences gathered from GenBank) for these species. Consequently, there is often no way to follow-up/recheck on questionable identifications (Floyd et al. Citation2010). The adoption of barcoding data standards for future collections (CitationHanner 2005), with at least the recording of photographic images and Global Positioning System (GPS) coordinates of capture for all reference specimens, would enhance the value of sequence data subsequently generated and this is a major objective of Fish Barcode of Life (FISH-BOL). Given the lack of vouchers for the billfishes, pooling all available samples and analyzing them with a common set of markers is a logical first step. However, we cannot rule out the possibility that some of the sequences, particularly those gleaned from GenBank, might derive from misidentifications.
The lack of resolution of COI to discriminate half of the billfish species is notable given the relative utility of DNA barcoding based on this locus in other fish groups (Ward et al. Citation2008; Wong and Hanner Citation2008; Steinke et al. Citation2009a,Citationb). Presumably the explanation lies in the fact that all of these species are relatively young in evolutionary time, and consequently there has been insufficient time for the accumulation of mutations. Speciation is a phenomenon which occurs at the interface between population genetic (reticulate) and phylogenetic (bifurcating) processes; species are diagnosable to the extent that different character states (alleles) become fixed in reproductively isolated populations, but the fixation of neutral alleles is a probabilistic process occurring over many generations (Padial et al. Citation2010), at heterogeneous rates for different genetic loci. This is particularly important in which genetic variation is neutrally evolving, and uncoupled to whatever phenotypic changes are responsible for species differences. In such cases, haplotypes deriving from the common ancestor population may persist for an indeterminate period of time in descendent populations, even after they have become fully reproductively isolated. As Padial et al. (Citation2010) emphasize, modern taxonomy must be integrated with many new approaches for species delimitation, and in such cases, species should be diagnosed by differing allele frequencies rather than by distinct allele sequences, which the generation of broader-coverage barcode data could help to reveal (Hajibabaei et al. Citation2007).
The three spearfishes T. angustirostris, T. belone, and T. pfluegeri could not be resolved based on our data (COI and Rho), even though all of these species were distinguishable based on the dataset employed by Collette et al. (Citation2006); this study used a concatenated suite of mitochondrial genes (ND2, CR, and 12S) and one nuclear gene (MN32), and found that these three species all formed reciprocally monophyletic groups with >70% bootstrap support. This is most likely a data sufficiency problem, i.e. simply the amount of genomic data which needs to be read to capture enough variation; the total length of concatenated sequence used by Collette et al. (Citation2006) was 3787 bp for each specimen, compared with 1112 bp for our study (length of COI+Rho sequences combined). Hence, some examples of barcoding missing ‘young’ species (Moritz and Cicero Citation2004) are expected, but in general, this had not proven to be an issue for the vast majority of fishes, both freshwater and marine, examined to date (Ward et al. Citation2009).
As an identification tool for an unknown tissue sample, barcoding using COI alone may be expected to provide at least a genus-level identification of billfish in all cases. On the data in this study, for half of the possible billfish species this will also yield an unambiguous species identification, whereas for the other half it will be necessary to sequence alternative genes to unambiguously determine the species. Thus, barcoding can provide a routine and effective ‘first pass’ for analysis of unknown samples, which can be refined by further sequencing as required.
Supplementary Material
Download (236.8 KB)Supplementary Material
Download JPEG Image (924.4 KB){kind=link}
Supplementary Material
Download PDF (281.9 KB)Supplementary Material
Download (34.7 KB)Supplementary Material
Download MS Excel (99 KB)Declaration of interest: This work was supported through funding to the Canadian Barcode of Life Network from Genome Canada (through the Ontario Genomics Institute), NSERC, and other sponsors listed at http://www.BOLNET.ca; and additionally by operational funds to the Guy Harvey Research Institute, AFTCO, Inc., and an NSERC postgraduate fellowship (AB). We thank L. Beerkircher, J. Graves, J. McDowell, E. Prince, J. Serafy, and D. Snodgrass for providing tissue samples and Eugene Wong for assistance in the laboratory. We are grateful to Johanne Fischer of the FAO for granting permission to use images from their catalog (CitationNakamura 1985) for the first figure in our study. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Arnot DE, Roper C, Bayoumi RAL. 1993. Digital codes from hypervariable tandemly repeated DNA sequences in the Plasmodium falciparum circumsporozoite gene can genetically barcode isolates. Mol Biochem Parasitol. 61:15–24.
- Clement M, Posada D, Crandall KA. 2000. TCS: A computer program to estimate gene genealogies. Mol Ecol. 9:1657–1659.
- Collette BBReproduction and development in epipelagic fishes. In: Cole K. editors. Reproduction and Sexuality in Marine Fishes: Patterns and Processes. Berkeley: University of California Press(2010)21–63 Chapter 2.
- Collette BB, McDowell JR, Graves JE. 2006. Phylogeny of Recent billfishes (Xiphioidei). Bull Mar Sci. 79:455–468.
- Costa FO, Carvalho GR. 2007. The Barcode of Life Initiative: Synopsis and prospective societal impacts of DNA barcoding of fish. Genom Soc Policy. 3:29–40.
- Ekrem T, Willassen E, Stur E. 2007. A comprehensive DNA sequence library is essential for identification with DNA barcodes. Mol Phylogenet Evol. 43:530–542.
- Floyd R, Abebe E, Papert A, Blaxter M. 2002. Molecular barcodes for soil nematode identification. Mol Ecol. 11:839–850.
- Floyd R, Lima J, deWaard J, Humble L, Hanner R. 2010. Common goals: Policy implications of DNA barcoding as a protocol for identification of arthropod pests. Biol Invasions. 12:2947–2954.
- GAO. 2009. Seafood fraud: FDA program changes and better collaboration among key federal agencies could improve detection and prevention. Report to the Ranking Member, Subcommittee on Oceans, Atmosphere, Fisheries, and Coast Guard, Committee on Commerce, Science, and Transportation, U.S. Senate, United States Government Accountability Office, #GAO-09-258, 53 pp.
- Gentner B. 2007. Economic analysis of international billfish markets: Executive summaryGentner Consulting Group8SilerSrgsMDavailableat http://www.gentnergroup.com/wp-content/uploads/final073107.pdf.
- Graves JE. 1998. Molecular insights into the population structures of cosmopolitan marine fishes. J Heredity. 89:427–437.
- Graves JE, McDowell JR. 2006. Genetic analysis of white marlin (Tetrapturus albidus) stock structure. Bull Mar Sci. 79:469–482.
- Hajibabaei M, Singer GA, Hebert PD, Hickey DA. 2007. DNA barcoding: How it complements taxonomy, molecular phylogenetics and population genetics. Trends Genet. 23:167–172.
- Hanner R. 2005. Proposed standards for BARCODE records in INSDC (BRIs), available at http://www.barcoding.si.edu/PDF/DWG_data_standards-Final.pdf on 2006.
- Hebert PDN, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc R Soc Lond B-Biol Sci. 2003a; 270:313–321.
- Hebert PDN, Ratnasingham S, deWaard JR. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B-Biol Sci. 2003b; 270 Suppl: S96–S99.
- Hsieh H-S, Chai T-J, Hwang D-F. 2005. Rapid PCR-RFLP method for the identification of 5 billfish species. J Food Sci. 70:246–249.
- Hsieh H-S, Chai T-J, Hwang D-F. 2007. Using the PCR-RFLP method to identify the species of different processed products of billfish meats. Food Control. 18:369–374.
- Hyde JR, Lynn E, Humphreys RJr, Musyl M, Vetter R. 2005. Shipboard identification of fish eggs and larvae by multiplex PCR, and description of fertilized eggs of blue marlin, shortbill spearfish, and wahoo. Mar Ecol Progress Ser. 286:269–277.
- Ivanova NV, Dewaard JR, Hebert PDN. 2006. An inexpensive, automation-friendly protocol for recovering high-quality DNA. Mol Ecol Notes. 6:998–1002.
- Ivanova NV, Zemlak TS, Hanner RH, Hebert PDN. 2007. Universal primer cocktails for fish DNA barcoding. Mol Ecol Notes. 7:544–548.
- Kawakami T, Aoyama J, Tsukamoto K. 2010. Morphology of pelagic fish eggs identified using mitochondrial DNA and their distribution in waters west of the Mariana Islands. Environ Biol Fishes. 87:221–235.
- Kumar S, Nei M, Dudley J, Tamura K. 2008. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 9:299–306.
- Luthy SA, Cowen RK, Serafy JE, McDowell JR. 2005. Toward identification of larval sailfish (Istiophorus platypterus), white marlin (Tetrapturus albidus) and blue marlin (Makaira nigricans) in the western North Atlantic Ocean. Fishery Bull. 103:574–587.
- McDowell JR, Graves JE. 2002. Nuclear and mitochondrial DNA markers for specific identification of istiophorid and xiphiid billfishes. Fish Bull. 100:537–544.
- McDowell JR, Graves JE. 2008. Population structure of striped marlin (Kajikia audax) in the Pacific Ocean based on analysis of microsatellite and mitochondrial DNA. Can J Fish Aquatic Sci. 65:1307–1320.
- Mora C, Myers RA, Coll M, Libralato S, Pitcher TJ, Sumaila RU, Zeller D, Watson R, Gaston K, Worm B. 2009. Management effectiveness of the world's marine fisheries. PLoS Biol. 7 6: e1000131 doi:10.1371/journal.pbio.1000131.
- Moritz C, Cicero C. 2004. DNA barcoding: Promise and pitfalls. PLoS Biol. 2 10: e354.
- Nakamura I. 1985. FAO species catalogue, Vol. 5: Billfishes of the World. An annotated and illustrated catalogue of marlins, sailfishes, spearfishes and swordfishes known to date. FAO Fisheries Synopsis Food and Agriculture Organization of the United Nations, #125, 65 pp.
- Nelson J. 2006. Fishes of the World. Hoboken, NJ: Wiley.
- Nicholas KB, Nicholas HBJ. 1997. GeneDoc: A tool for editing and annotating multiple sequence alignments. Distributed by the author, http://www.psc.edu/biomed/genedoc.
- Padial J, Miralles A, De la Riva I, Vences M. 2010. The integrative future of taxonomy. Front Zool. 7:1–14 doi:10.1186/1742-9994-7-16.
- Ratnasingham S, Hebert PDN. 2007. The Barcode of Life Data Systems (www.barcodinglife.org). Mol Ecol Notes. 7:355–364.
- Richardson DE, Vanwye JD, Exum AM, Cowen RK, Crawford DL. 2007. High-throughput species identification: From DNA isolation to bioinformatics. Mol Ecol Notes. 7:199–207.
- Richardson DE, Llopiz JK, Guigand CM, Cowen RK. 2010. Larval assemblages of large and medium-sized pelagic species in the Straits of Florida. Progress Oceanogr. 86:8–20.
- Sedberry G, Loefer J. 2001. Satellite telemetry tracking of swordfish, Xiphias gladius, off the eastern United States. Mar Biol. 139:355–360.
- SEFSC/NMFS. 2004. Atlantic Billfish Research Plan. National Marine Fisheries Service. Miami, FL.: South East Fisheries Science Center, National Oceanic and Atmospheric Administration 44 pp. available at: http://www.sefsc.noaa.gov/docs/ABRP_01_30_04.pdf.
- Sevilla RG, Diez A, Noren M, Mouchel O, Jerome M, Verrez-Bagnis V, Van Pelt H, Favre-Krey L, Krey G, Bautista J, The Fish Trace Consortium. 2007. Primers and polymerase chain reaction conditions for DNA barcoding teleost fish based on the mitochondrial cytochrome b and nuclear rhodopsin genes. Mol Ecol Notes. 7:730–734.
- Shivji MS, Magnussen JE, Beerkircher LR, Hinteregger G, Lee D, Serafy J, Prince E. 2006. Validity, identification, and distribution of the roundscale spearfish, Tetrapturus georgii (Teleostei: Istiophoridae): Morphological and molecular evidence. Bull Mar Sci. 79:483–492.
- Steinke D, Zemlak T, Boutillier J, Hebert P. DNA barcoding of Pacific Canada's fishes. Mar Biol. 2009a; 156:2641–2647.
- Steinke D, Zemlak TS, Hebert PDN. Barcoding nemo: DNA-based identifications for the ornamental fish trade. PLoS ONE. 2009b; 4 7: e6300 doi:10.1371/journal.pone.0006300.
- Victor BC, Hanner R, Shivji M, Hyde JR, Caldow C. 2009. Identification of the larval and juvenile stages of the cubera snapper, Lutjanus cyanopterus, using DNA barcoding. Zootaxa. 2215:24–36.
- Ward RD, Costa FO, Holmes BH, Steinke D. 2008. DNA barcoding of shared fish species from the North Atlantic and Australasia: Minimal divergence for most taxa, but Zeus faber and Lepidopus caudatus each probably constitute two species. Aquatic Biol. 3:71–78.
- Ward RD, Hanner R, Hebert PDN. 2009. The campaign to DNA barcode all fishes, FISH-BOL. J Fish Biol. 74:329–356.
- Wong EH-K, Hanner RH. 2008. DNA barcoding detects market substitution in North American seafood. Food Res Int. 41:828–837.
- Yancy HF, Zemlak TS, Mason JA, Washington JD, Tenge BJ, Nguyen NT, Barnett JD, Savary WE, Hill WE, Moore MM, Fry FS, Randolph SC, Rogers PL, Hebert PDN. 2008. Potential use of DNA barcodes in regulatory science: Applications of the regulatory fish encyclopedia. J Food Protect. 71:210–217.