
Abstract
Our objective was to provide demographic profiles and incidence estimates of amyotrophic lateral sclerosis (ALS) in two diverse California metropolitan areas: Los Angeles County (LA) and the San Francisco Bay Area (SFBA). Data were retrospectively collected from multiple sources. Case eligibility criteria included residency in SFBA or LA, and treatment for or diagnosis of ALS between 1 January 2009 and 31 December 2011. Overall incidence rates as well as age-, gender-, race- and ethnicity-specific rates were calculated. We identified 539 ALS cases in SFBA and 545 in LA; 618 were incident cases. Cases were more likely to be male and white. There were considerably more cases (p < 0.05) in LA who were foreign-born (LA, 22%; SFBA, 15%), black (LA, 10%; SFBA, 6%) or Hispanic (LA, 19%; SFBA, 10%). Conversely, the age adjusted incidence rates (per 100,000) were higher in SFBA for whites (LA, 1.40; SFBA, 2.49) and Hispanics (LA, 0.66; SFBA, 1.57) compared with LA. General case demographics and incidence rates in these two areas were similar to published studies. However, the differences between the two areas raise questions about how factors such as geography, access to care, and referral patterns may affect case ascertainment and diagnosis.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is the most common motor neuron disease and is characterized by progressive deterioration of upper and lower motor neurons. ALS is age related, with the highest rate of onset between 55 and 75 years (Citation1,Citation2). The average survival time after symptom onset is 2–5 years; only a small percentage of patients survive beyond five years (Citation1,Citation2). Males have a slightly higher prevalence than females (Citation1–3). Little is known about the etiology of ALS; only 5–10% can be attributed to family history, leaving many questions about possible environmental factors (Citation1–4).
The annual incidence rate of ALS in the United States is estimated to be 1.6–2.0 per 100,000 population (Citation5). Subpopulation incidence and prevalence rates for ALS are difficult to find due to the lack of representative data. The majority of ALS epidemiologic studies have focused on racially and ethnically homogenous population samples in Europe and non-Western countries. Among these, there is relatively uniform incidence among white populations and a lower incidence among African, Asian, and Hispanic populations. However, it is difficult to compare ALS estimates across these studies and populations due to different case-finding methodologies (Citation1,Citation6–12).
In 2008, the U.S. Congress mandated that the Centers for Disease Control and Prevention (CDC) establish the National ALS Registry with the primary goal of determining national incidence and prevalence rates for ALS (Citation13). The CDC's sister public health agency, the Agency for Toxic Substances and Disease Registry (ATSDR), created and maintains the registry. To validate the completeness and accuracy of registry data, ATSDR funded three state and eight metropolitan-area surveillance projects. This manuscript focuses on ATSDR's collaboration with the California Environmental Health Tracking Program – at the California Department of Public Health – to conduct metropolitan-area surveillance projects in Los Angeles County (LA) and the San Francisco Bay Area (SFBA). Site- specific goals included obtaining incidence rates for LA and SFBA and improving understanding of the demographic characteristics of the ALS population.
Materials and methods
We conducted retrospective surveillance to identify ALS cases in SFBA and LA who were seen or diagnosed by a neurologist between 1 January 2009 and 31 December 2011. The project population consisted of residents from two metropolitan areas: five SFBA counties (Alameda, Contra Costa, San Mateo, San Francisco, and Solano Counties) in Northern California with a combined population of 4.5 million and LA County in Southern California with a population of 9.8 million. These areas were chosen for their racial and ethnic diversity. According to the U.S. Census (Citation14), SFBA counties were 23% Asian, 9% black, and 50% white, while LA was 14% Asian, 9% black, and 50% white. Among these, 22% and 48% were Hispanic (or any race), respectively. Both areas were 51% female and 49% male.
Case ascertainment
Primary data collection. A comprehensive list of neurologists was compiled from physician lists provided by the ALS Association (ALSA), state medical board, and internet searches. ALSA facilitated initial contacts with ALS care centers and large referral centers. An ALS care center was defined as a specialty center if it was funded by the ALSA and the Muscular Dystrophy Association, and large referral centers are practices that diagnosed or provided care for at least 50 ALS patients during the reporting period. Physicians from smaller practices were identified and contacted by project staff through mailings, phone calls, faxes, and office visits.
Project staff contacted practicing neurologists serving the catchment areas to determine whether ALS patients were treated in their facility, and how many patients met the case definition. Neurology office staff identified potential cases by searching electronic billing records, patient databases, or directly consulting with the neurologist. Patients were reported if they could be classified into one of the El Escorial criteria categories (Citation15) by the neurologist using the patients’ medical record. In some instances where the reporting neurologist felt strongly that a patient had ALS but could not be classified into one the El Escorial criteria categories, these patients were placed into the unclassifiable category (Citation16).
Secondary data collection. Using California's Electronic Death Registration System (EDRS), project staff obtained records of all deaths among residents of SFBA or LA between 1 January 2009 and 31 December 2011. Staff identified decedents with ALS as one of the causes of death who had not been captured through primary data collection. Attending physicians listed on the death record were contacted to verify diagnosis and obtain an ALS case report.
Case definition
ALS cases meeting the El Escorial criteria, who resided in SFBA or LA and who were diagnosed or under the care of a neurologist between 1 January 2009 and 31 December 2011, were eligible for reporting. Residence was determined by zip code of record.
Data collection and quality assurance
Office staff completed a standardized case report form by abstracting case demographics – race, ethnicity, country of birth, address, diagnosis, dementia status, and family history of ALS – from patient charts. Identifying data elements such as name, date of birth, and partial social security number (SSN) were collected to de-duplicate cases and to compare with the National ALS Registry data. We followed U.S. federal guidelines and recorded race and ethnicity separately (Citation17). Patients with more than one race indicated in their medical charts were classified as mixed race. Date of diagnosis was the first date a neurologist noted the patient had ALS in the medical chart. Data were entered into a Microsoft Access database and ongoing quality control checks were conducted to ensure case reports and electronic data matched. Cases that matched others based on name, SSN, or date of birth were flagged for review. Duplicate reports and death data, when available, were merged to create complete data on each case as necessary. A unique record was created for each individual; duplicate reports were not included in the final datasets.
A sample, of approximately 15% of cases, was systematically selected for quality assurance. For the selected cases, neurologists were asked to complete a more detailed medical records verification form and submit a recent electromyogram (EMG) to ensure diagnostic accuracy. To verify diagnosis, the completed forms and EMGs were reviewed by a consulting neurologist who specialized in the diagnosis and treatment of ALS. The consulting neurologist was blinded to the practice and neurologist reporting the case.
Statistical analysis
Age at symptom onset and diagnosis and time from symptom onset to diagnosis were calculated for complete records. χ2 or Fisher's tests as appropriate were used to assess differences between groups and one-way analysis of variance (ANOVA) to compare continuous variables. A p-value < 0.05 was considered significant.
Incident cases
Age adjusted incidence estimates were calculated by site (SFBA v. LA) including only cases with dates of initial diagnosis occurring from 1 January 2009 to 31 December 2011. Age-specific, race, and ethnicity estimates were also calculated by site. For all rate calculations, cases and populations were stratified by 10-year age groups, with those older than 80 years of age constituting a single group; rates were standardized using the US 2000 Census population. Age-adjusted rates and confidence intervals were calculated using the protocol of Tiwari, Clegg, and Zhou (Citation18), while standardized rate ratios between the LA and SFBA areas were calculated using the method found in Newman (Citation19).
Data cleaning and statistical analyses were conducted using Microsoft Excel and SAS v9.3 (Cary NC) (Citation20). This project was approved by the Center for Disease Control and Prevention Institutional Review Board (IRB) and determined to be public health surveillance (rather than human subjects research), and therefore not requiring institutional review board review, by the California Committee for the Protection of Human Subjects.
Results
Using multiple data sources, we identified 95% of expected cases (Citation5). A disproportionate number were from SFBA, where we identified 150% of expected cases (539/360) compared with LA where we identified 69% (545/785). Over half (57%) of the cases were incident cases with initial diagnosis occurring between 2009 and 2011 (SFBA, 288; LA, 330). All 12 of the ALS care centers (four in SFBA and six in LA), and large referral centers (one in SFBA and one in LA) in both catchment areas participated, and 83% (77/93) of practices that reported seeing ALS patients submitted case reports.
Characteristics of reported cases
The distributions of ALS cases by gender, race, ethnicity, and US-born status for SFBA, LA, and combined areas are shown in . Males represented the majority of the cases – 320 (59%) and 302 (55%) in SFBA and LA, respectively. White cases made up 72% (390) in SFBA and 70% (379) in LA. LA had twice as many Hispanic cases as SFBA – 102 (19%) vs. 55 (10%). Both sites had substantial yet differing proportions of cases with race or ethnicity not reported (). There were no differences in the percentage of cases with health insurance by geographic area; 98% of patients had at least one type of insurance (data not presented). There were significantly more foreign-born cases reported in LA (22%) than in SFBA (15%). Country of birth was missing in 25% of cases overall.
Table I. Characteristics of ALS cases identified for San Francisco Bay Area (SFBA) and Los Angeles County (LA), 2009–2011.
Mean age at diagnosis was 62.2 years (range 14–94 years) in SFBA and 60.8 years (range 14–90 years) in LA. There was a significant difference in the age of symptom onset between sites: 60.6 (range 13 –94) years in SFBA and 58.8 (range 11–90) years in LA. Mean time from symptom onset to diagnosis was 21 months (range 0–36 months), while the median time was 12 months.
Familial history of ALS was indicated in 3% (Citation17) and 4% (Citation22) of cases in SFBA and LA, respectively. Cases with a diagnosis of dementia were reported in 34 (6%) in SFBA and 22 (4%) in LA.
Characteristics of incident cases
There were 618 (SFBA, 288; LA, 330) patients diagnosed between 1 January 2009 and 31 December 2011. The distribution of gender, race, ethnicity, country of birth, family history of ALS, and presence of a dementia diagnosis was similar to the full set of reported cases ().
Among these cases, overall mean age at time of diagnosis was 63.7 years (range 17–92 years), with 65.2 years (range 32–92 years) in SFBA and 62.4 years (range 17–90 years) in LA. Overall mean age at symptom onset was 61.9 years (range 17–92 years); 63.7 (range 28–92 years) in SFBA and 60.4 years (range 17–90 years) in LA.
Incidence rate calculations
Overall crude incidence rate was 1.4 per 100,000 (SFBA and LA: 2.1 and 1.1, respectively, per 100,000). Overall age-adjusted incidence rate was 1.7 per 100,000. The age-adjusted incidence estimate was higher in SFBA (2.0; CI = 1.8–2.3) compared with LA (1.2; CI = 1.0–1.3).
Age distribution of incident ALS was similar in both catchment areas, increasing with age until 80 years with the highest incidence among the 70–79 years age group (SFBA and LA: 11.7 and 5.9, respectively) (). Based on standardized rate ratios (SRRs), differences in age-specific incidence rates between SFBA and LA were largest among the older age groups ().
Figure 1. Age-specific incidence of ALS, SF Bay Area and Los Angeles counties, 2009–2011.
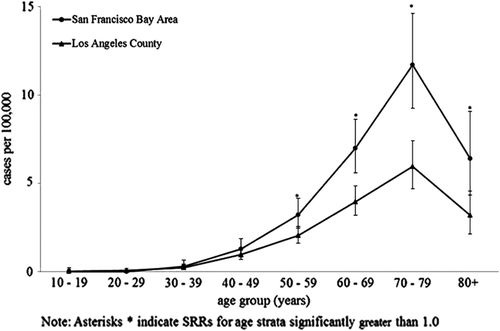
Table II. Age-standardized incidence estimates and standardized rate ratios (SRR) for San Francisco Bay Area (SFBA) and Los Angeles County (LA), 2009–2011.
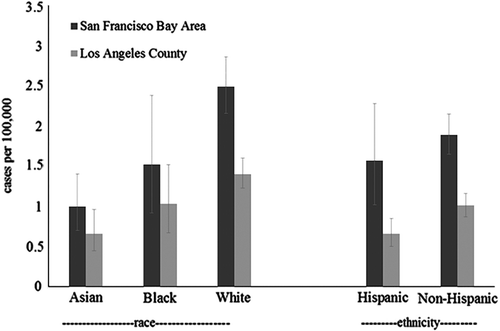
Within SFBA, whites had a notably higher incidence (2.5; CI = 2.2–2.9) than Asians (1.0; CI = 0.7–1.4), with the rate for blacks lying between these two (1.5; CI = 0.9–2.4). This pattern was similar in LA, with the rate among whites 1.4, Asians 0.7, and blacks 1.0. In each area, estimates were slightly lower among Hispanics compared to non-Hispanics (SFBA: 1.6 (1.0–2.3) versus 1.9 (1.7–2.2)); LA: 0.7 (0.5–0.9) versus 1.0 (0.9–1.2)).
Quality assurance
One hundred sixty-four cases were selected for review by the consulting ALS specialist for diagnosis verification: 69% from ALS care and referral centers and 31% from other practices. All of the cases reviewed from the unclassifiable category were found to meet the El Escorial criteria for ALS. Among all the reviewed cases, four (2.4%) were classified as not ALS and excluded from the final case count.
Discussion
This surveillance exercise presents data on ALS patients from two diverse metropolitan areas using multiple data sources. The size and diversity of the sample allowed us to describe the epidemiology of ALS cases and to provide the most thorough estimates – with some limitations – to date in CA, specifically for SFBA and LA.
Demographic characteristics among identified cases such as age of onset, slightly higher proportion of males, and higher proportion of white cases are similar to existing literature (Citation1–3). The percentage of familial ALS reported in this project ranged from 3% to 4%, which is slightly lower than the 5–10% of familial ALS frequently reported in the literature (Citation4). In Chio et al.'s review, the mean reported time from onset to diagnosis was 9–15 months (Citation1); our project found a much longer delay at 21 months. Among studies reporting the median, the time between onset and diagnosis was 9–12 months, which is similar to what was observed in our project (12 months) (Citation3,Citation21–24).
Incidence
The overall age-adjusted incidence rate is comparable to previously estimated rates in the U.S. and lower than the rates reported in Logroscino et al. (Citation5,Citation10,Citation24–29). Incidence rates among white populations have consistently been higher than non-white; this likely explains the differences between the rates reported in Europe and the rates reported in the U.S. where a higher percentage of the population is non-white. The lower incidence rate could also be related to this project's retrospective design, which has been suggested can result in lower estimates compared to prospective studies (Citation1). The European studies were largely prospective utilizing data from established registries (Citation16,Citation30–32).
Incidence rates increased with age, peaking at 70–79 years followed by a marked decline. This follows the general pattern found in Logroscino's pooled analysis of six European registries (Citation33) and a recent systematic review of population based studies published globally (Citation1).
Within our project, SFBA had a higher incidence rate than LA. Incidence of white cases was higher, and we found differences among specific race/ethnicities by region. Previous research has found conflicting results in racial and ethnic differences in national ALS incidence rates (Citation6,Citation24,Citation25,Citation34). We found that Asians had the lowest rates and had notably lower rates than whites in both regions. No differences were found between the incidence rates of whites and blacks, and Hispanics had a lower incidence in LA only.
It is important to understand the effect that missing data have on our race- and ethnicity-specific rate estimates (, and ). Because there is not an ‘unknown’ category for race or ethnicity in the census data, our denominator values can be considered to be a more complete representation of populations in each racial or ethnic category than can our numerator values. Therefore, compared to our population-wide incidence estimates, our subgroup incidence estimates should be considered downwardly biased, particularly regarding ethnicity, and particularly for LA. Race- and ethnicity-specific SRRs are more difficult to interpret because of higher rates of missing data in LA, which biases these estimates upwards.
Figure 2. Age-adjusted race and ethnicity specific incidence of ALS, SF Bay Area and Los Angeles counties, 2009–2011.
The large number of cases with unknown race and ethnicity can partly be attributed to differences in the collection and categorization of race and ethnicity across providers (who often combined race and ethnicity) compared with the categories we used in our project (which separate race and ethnicity). With additional information unavailable, chart abstractors were not always able to fill in separate responses for race and ethnicity. Although the U.S. federal standards (Citation17) state that Hispanic origin (ethnicity) and race (regardless of ethnicity) are distinct groupings and should be collected separately, these standards are not mandated outside of federal agencies and federally supported projects.
Different referral patterns were observed between sites. For example, in the SFBA, the majority of neurology practices referred suspected cases to the ALS care centers (all of which reported cases), while in LA fewer practices made these referrals, resulting in more practices to follow up with, and fewer patients with records maintained in centralized electronic record systems. This referral pattern may have disproportionately affected Hispanics in LA, where a larger percentage were foreign-born, and additional factors – such as language, culture and access – may have impacted the likelihood of seeking care at an ALS center. Reasons for the greater than expected number of cases in SFBA and the fewer than expected in LA are unclear; the differences may represent true incidence rates in populations with varying sociodemographics and unidentified risk factors. However, the differences may be a result of dissimilarities in health care systems, rates of insurance coverage, and/or access to medical care in the two distinct geographic regions. Further investigation into factors affecting case ascertainment is warranted.
Strengths and limitations
Two primary limitations should be considered when interpreting the findings: first, the likely under-ascertainment of ALS cases, which may have affected rate estimates for LA, particularly among Hispanics. Secondly, incomplete data on race/ethnicity may have underestimated subgroup incidence rates. This project also has several strengths. The intensive case ascertainment process yielded a high number of confirmed cases from a variety of clinical settings, unique to ALS surveillance efforts. Quality assurance methods were used to assure that neurologists were assigning the El Escorial criteria appropriately. Finally, analyses and incidence rates presented here allow for comparisons with similar studies and fill gaps in the literature.
Conclusions
In summary, we collected ALS case data from two metropolitan areas with ethnically diverse populations. These data suggest that overall ALS incidence appears to be comparable to recent publications. Future studies are needed to assess whether the demographic patterns found in our surveillance data represent true disease patterns.
Moreover, the differences in case ascertainment could inform future surveillance efforts by the National ALS Registry. The forthcoming results of the national ALS surveillance effort (of which this project is one component) will be an important next step for better understanding the current epidemiology of ALS among the U.S. population.
Acknowledgements
We would like to thank Eric Sorenson for evaluating and verifying case diagnoses. This project was funded by McKing Consulting Corporation through a contract funded by the federal Agency for Toxic Substances and Disease Registry.
Declaration of interest: Wendy Kaye has received personal compensation for activities with McKing Consulting Corporation as an employee.
The conclusions of this article are those of the authors and do not necessarily represent the views of ATSDR, CDC, or the U.S. Department of Health and Human Services.
References
- Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology. 2013;41:118–30.
- Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet Journal of Rare Diseases. 2009;4:3.
- Logroscino G, Traynor B, Hardiman O, Couratier P, Mitchell J, Swingler R, et al. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. Journal of Neurology, Neurosurgery & Psychiatry. 2008;79:6–11.
- Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82:623–7.
- Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the ‘common’ neurologic disorders? Neurology. 2007;68:326–37.
- Cronin S, Hardiman O, Traynor BJ. Ethnic variation in the incidence of ALS: a systematic review. Neurology. 2007;68:1002–7.
- Dietrich-Neto F, Callegaro D, Dias-Tosta E, Silva HA, Ferraz ME, Lima J, et al. Amyotrophic lateral sclerosis in Brazil: 1998 national survey. Arquivos de Neuro-psiquiatria. 2000;58:607–15.
- Doi Y, Yokoyama T, Tango T, Takahashi K, Fujimoto K, Nakano I. Temporal trends and geographic clusters of mortality from amyotrophic lateral sclerosis in Japan, 1995–2004. J Neurol Sci.2010;298:78–84.
- Forsgren L, Almay BG, Holmgren G, Wall S. Epidemiology of motor neuron disease in northern Sweden. Acta neurologica Scandinavica. 1983;68:20–9.
- Sorenson EJ, Stalker AP, Kurland LT, Windebank AJ. Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925 to 1998. Neurology. 2002;59:280–2.
- Kazamel M, Cutter G, Claussen G, Alsharabati M, Oh SJ, Lu L, et al. Epidemiological features of amyotrophic lateral sclerosis in a large clinic-based African-American population. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:334–7.
- Zaldivar T, Gutierrez J, Lara G, Carbonara M, Logroscino G, Hardiman O. Reduced frequency of ALS in an ethnically mixed population: a population based mortality study. Neurology. 2009;72:1640–5.
- Antao VC, Horton DK. The National Amyotrophic Lateral Sclerosis (ALS) Registry. Journal of Environmental Health. 2012;75:28–30.
- 2010 Demographic Profile by County [Internet]. U.S. Census Bureau, 2010 Census. [cited 2013]. Available from: http://factfinder2.census.gov/.
- Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2000;1:293–9.
- Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: a population based study. Arch Neurol. 2000;57:1171–6.
- Office of Budget and Management. Revisions to the Standards for the Classification of Federal Data on Race and Ethnicity. Federal Register. 1997;62:58781–90.
- Tiwari RC, Clegg LX, Zou Z. Efficient interval estimation for age-adjusted cancer rates. Statistical Methods in Medical Research. 2006;15:547–69.
- Newman SC. Biostatistical methods in epidemiology: John Wiley & Sons; 2003.
- SAS Institite Inc. Base SAS® 9.3. Cary, NC: SAS Institute Inc.
- Cellura E, Spataro R, Taiello AC, La Bella V. Factors affecting the diagnostic delay in amyotrophic lateral sclerosis. Clinical Neurology and Neurosurgery. 2012;114:550–4.
- Murphy M, Quinn S, Young J, Parkin P, Taylor B. Increasing incidence of ALS in Canterbury, New Zealand: a 22-year study. Neurology. 2008;71:1889–95.
- Werneck LC, Bezerra R, Silveira Neto Od*, Scola RH. A clinical epidemiological study of 251 cases of amyotrophic lateral sclerosis in the south of Brazil. Arquivos de Neuro-Psiquiatria. 2007;65:189–95.
- Jordan H, Fagliano J, Rechtman L, Lefkowitz D, Kaye W. Population-Based Surveillance of Amyotrophic Lateral Sclerosis in New Jersey, 2009–2011. Neuroepidemiology. 2014;43:49–56.
- Annegers JF, Appel S, Lee JR-J, Perkins P. Incidence and prevalence of amyotrophic lateral sclerosis in Harris County, Texas, 1985–1988. Arch Neurol. 1991;48:589–93.
- Turabelidze G, Zhu B-P, Schootman M, Malone JL, Horowitz S, Weidinger J, et al. An epidemiologic investigation of amyotrophic lateral sclerosis in Jefferson County, Missouri, 1998–2002. Neurotoxicology. 2008;29:81–6.
- Report of the Steering Committee to Reengineer the Death Registration Process. Toward an electronic death registration system in the United States. Am J Forensic Med Pathol. 1998;19:234–41.
- Wagner L, Archer N, Williamson D, Henry J, Schiffer R, Jackson C. Prevalence of amyotrophic lateral sclerosis in Texas, 1998–2003. Tex Med. 2012;108:e1.
- McGuire V, Longstreth W, Koepsell TD, van Belle G. Incidence of amyotrophic lateral sclerosis in three counties in western Washington state. Neurology. 1996;47:571–3.
- Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10:310–23.
- Logroscino G, Beghi E, Zoccolella S, Palagano R, Fraddosio A, Simone I, et al. Incidence of amyotrophic lateral sclerosis in southern Italy: a population based study. Journal of Neurology, Neurosurgery & Psychiatry. 2005; 76:1094–8.
- Beghi E, Millul A, Micheli A, Vitelli E, Logroscino G. Incidence of ALS in Lombardy, Italy. Neurology. 2007; 68:141–5.
- Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, et al. Incidence of amyotrophic lateral sclerosis in Europe. Journal of Neurology, Neurosurgery & Psychiatry. 2010;81:385–90.
- Matsumoto N, Worth RM, Kurland LT, Okazaki H. Epidemiologic study of amyotrophic lateral sclerosis in Hawaii: identification of high incidence among Filipino men. Neurology. 1972;22:934–40.