
Abstract
Lipid vesicles are an important drug carrier which can serve for controlled delivery of drugs; however, these vesicles are quite unstable at ambient temperature and require stringent storage condition.
Present work was done to develop a stable vesicular system for drug delivery. Vesicles of ceramide-2, cholesterol, cholesterol sulfate, and palmitic acid were prepared and compared with phosphatidylcholine vesicles for physicochemical parameters and accelerated stability. Diclofenac sodium was used as a model drug.
Based on physicochemical parameter and in vitro release PCV-3 and CV-3 were selected for further studies in three different accelerated stability conditions. PCV-3 showed moderate changes at 4°C but was severely affected at 25°C and 40°C. CV-3 showed stable characteristics at 4°C and 25°C whereas at 40°C, CV-3 showed signs of slight modification owing to moisture absorption.
Based on the study, CV-3 containing highest content of palmitic acid was found to be most stable.
Introduction
Development of the delivery system is of utmost importance to improve the therapeutic efficacy of newly developed drug candidates as well as for new indications of pre-existing drugs. The need of the hour is to provide controlled delivery of drug to the specific site of action. Several types of drug carriers have been developed owing to the peculiar requirements of a site of drug administration; however, every carrier has one or other limitation (Kostarelos Citation2003, Gaur et al. Citation2014a). Liposomes are important examples of bio- membranes which have been successfully used as vehicles for sustained delivery. It offers several advantages over conventional drug carriers such as enhancing systemic exposure of the drug and can decrease the toxicity. It also improves the bioavailability of poorly soluble drugs and delays elimination of rapidly metabolized drugs. It can be used to deliver both hydrophilic and lipophilic drugs (Nasr et al. Citation2008, Blume and Cevc Citation1992, Zheng et al. Citation1992).
Structurally, liposomes comprise of an outer shell of one or more membrane of the molecules arranged concentrically around hollow interior of ‘vacuole‘ which serves as storage compartment for the active agent. Liposomes are predominantly removed from the body due to the action of reticulo-endothelial system. Liposomes can accommodate physicochemically different drugs in liposome membrane (Hydrophobic) and internal core (hydrophilic). The enhancement of solubility is around 100- to 10000-fold for liposome resulting in effective drug delivery (Li et al. Citation2007, Yadav et al. Citation2011). Since last three decades liposomes have been studied for various applications, but their stability remains the main concern. Vesicle formation is often a spontaneous process; however, it does not have equilibrium because non-equilibrium methods (e.g. sonication) are used for their formulation. These systems can again re-equilibrate back into multi-lamellar, liquid crystalline structures thereby showing physical instability. Further, the main component of conventional liposomes, phosphatidylcholine does not have good stability at physiological temperature giving rise to chemical instability in formulations (Li et al. Citation2007, Gaur et al. Citation2010, Yadav et al. Citation2011) so alternatives to phosphatidylcholine should be the focus from a technical view point to increase the stability of liposome formulations (Baillie et al. Citation1985).
Ceramides are natural lipid molecules which maintain hydrophobicity and physiological role of skin (Walters and Roberts Citation2002, Wertz et al. Citation1986, Gaur et al. Citation2014b). Presently ceramides have been used in anti-ageing cosmetic products and also as therapeutic agents in atopic dermatitis and xerosis by means of reduced trans- epidermal water loss (Weber et al. Citation2012, Lintner et al. Citation1997). Ceramides form bilayer and Goñi and Alonso have published a detailed review on membrane properties of ceramides (Goñi and Alonso Citation2006, Wartewig and Neubert Citation2007, Yardley and Summerly Citation1981).
Diclofenac is used in the study as a model drug. Diclofenac is an anti-inflammatory drug which generally produces gastrointestinal disturbances upon continuous usage.
This work was done to formulate a stable carrier for drug delivery. The developed vesicles were compared with conventional phosphatidylcholine vesicles in terms of physicochemical parameters, in vitro drug release and stability.
Materials and methods
Materials
Diclofenac Sodium and Phosphatidylcholine (PC) were kind gifts from Asoj Soft Caps, Baroda, India and Ceramide- 2 (N-stearoyl-(2S,2R,3R)-2-amino-1,3-octadecandiol or N-stearoyl-DL-sphinganin) was a kind gift from Sederma, France (Lot No.0000380664; patent No. US 2004-0120918) was supplied by Croda Chemicals (India) Pvt Ltd, Thane, Mumbai, India. Cholesterol (Chol) and Dicetyl phosphate (DCP) were procured from Himedia, Mumbai, India. Cholestryl-3-sulphate (CS), Hexadecanoic acid (Palmitic acid ≥ 99%, PA) were procured from Sigma–Aldrich (New Delhi, India).
Methods
Preparation of vesicles
Lipid film hydration method was used for liposome preparation. The composition is given in . Chloroform solution of drug was mixed with lipids in the predefined ratio. Then it was evaporated in round-bottom flask to make a film on inner surface of flask (Rotavapor® R II, BUCHI India Private Ltd.). The solvent traces were removed by applying vacuum for 1 h. The film was hydrated using acetate buffer solution (pH 5.5) at 200 rpm for 1 h at 25°C. The dispersion was then sonicated using a probe sonicator for 15 min at 100 W amplitude to obtain a homogeneous nanodispersion. The nanodispersion was extruded through a membrane (Immobilon-P Membrane, 0.22 μm pore size, Millipore Pvt. Ltd., New Delhi,India). Gel filtration by Sephadex G-20 minicolumn was used to remove unentrapped drug from liposomes (Sinico et al. Citation2005, Gaur et al. Citation2014b).
Table I. Lipid composition for different vesicle formulations (Molar ratio).
Determination of morphology and size of vesicles
Liposome's size and shape were determined using Transmission electron microscopy through negative staining. The sample was located over a 200-mesh copper grid and permitted to absorb. Then phosphotungstic acid (1%) was added and extra traces were removed. Then samples were analyzed using Philips CM-10 (Acceleration voltage: 100 kV; Magnification: upto 10,000×; Cryo-attachment).
Charge characteristics and polydispersity index
Poly-dispersity indices and zeta potential were determined using the Zetasizer (NanoZS, Malvern Instruments, UK). The samples were lyophilized and dispersed in pH 7.4 phosphate buffer for analysis.
Entrapment efficiency
Ultracentrifugation was used for analyzing entrapment efficiency. Aliquots of vesicles were subjected to centrifugation using ultracentrifuge (Remi C-24 BL with angular R-241 rotor, Remi House, Mumbai, India). The vesicles were centrifuged at 12,000 rpm and sediment and supernatant were analyzed for drug content (Touitou et al. Citation2000, Gaur et al. Citation2014b, An et al. Citation2011). The entrapment efficiency was calculated using following formula:
where T = total amount of drug (supernatant + sediment) and C = amount of drug in supernatant.
HPLC assay of diclofenac sodium
HPLC assay using Shim-pack VP-ODS, 4.6 mm I.D. × 150 mm, 5 μm particle size (Shimadzu, Japan) with solvent system: methanol/water/acetic acid (80:20:0.5, v/v/v) was used for drug content estimation (Baydoun and Muller-Goymann Citation2003, Liu et al. Citation2011, Gaur et al. Citation2014b). The injection volume, flow rate and detection wavelength were 20 μl, 1.0 ml/min and 275 nm, respectively. Calibration curve was prepared between the concentration range of 2–40 ng/ml. Calibration curve equation obtained was y = 11256x + 544.9 (R2 = 0.998), where x is the concentration and y is the peak area.
In vitro drug release
Synthetic cellulose acetate membrane with molecular cutoff 12 KDa was used for drug release study. Membrane was kept in physiological saline at 37 ± 0.5°C and was placed over Franz diffusion cells (3.14 cm2) having phosphate buffer saline (PBS) with pH 7.4 in acceptor compartment. Then, formulation (˜ 5 mg drug) was applied, aliquots were taken out till 24 h and drug content was estimated using HPLC assay (Dragicevic-Curic et al. Citation2008, Gaur et al. Citation2014b).
Stability studies
ICH guidelines were followed for stability determination. The samples were placed in stability chamber for 25 ± 0.5°C and 40 ± 0.5°C and in refrigerator for 4 ± 0.5°C. The physicochemical parameters were determined after 1, 2 and 3 months of storage for stability analysis (Alam et al. Citation2008, Ishida et al. Citation2002, Arifin and Palmer Citation2005).
Results and discussion
The study was designed to formulate a vesicular formulation with acceptable stability during ambient storage conditions. The idea was to modify the lipid composition to replace phosphatidylcholine with a more stable natural lipid. We used ceramide-2 with cholesterol, cholesterol sulfate and palmitic acid for formulating vesicles which were compared with phosphatidylcholine vesicles, made up with PC, cholesterol and dicetyl phosphate. Both formulations were evaluated under accelerated conditions for determining the stability.
Phosphatidylcholine is the major component of liposomal membrane. Composition is given in . Further the addition of cholesterol makes firm vesicle wall, since it is a flat molecule and seals the gaps between adjacent PC molecules. Dicetyl phosphate is used for incorporating negative charge in liposomal membrane. The inclusion of charge keeps the vesicles segregated and reduces coalescence. The phase transition temperature of PC is 41°C. At or above phase transition temperature, liquid state vesicles are formed while below the Tm, gel state vesicles are formed. PC is a neutral molecule since it carries both charges, positive on quaternary nitrogen and negative on phosphate group.
Ceramide-2 is the major component of ceramide vesicles. Composition is given in . Cholesteryl sulfate is the crucial component to formulate ceramide vesicles. Ceramide-2 carries only positive charge and is categorized in cationic lipids. It is found in stratum corneum where it forms a major portion of lipid barrier along with ceramide 5. The phase transition temperature of ceramide 2 is 61 C which is reduced to 46°C at physiological pH of skin (4.5) so it forms gel state vesicles (Moore and Rerek Citation2000).
Different formulations were made to optimize the lipid composition. All compositions yielded spherical vesicles. The TEM images for best PC vesicle and ceramide vesicle (PCV-3 and CV-3) have shown in .
Figure 1. TEM images of formulations (10000×); a: PC vesicle (PCV-3); b: Ceramide vesicle (CV-3). (a) PC vesicle (PCV-3); (b) Ceramide vesicle (CV-3).

PCV-1 showed largest vesicle size and higher polydispersity index which means this composition is not suitable for making liposomes. PCV-2 showed moderate vesicle size and PDI and most negative zeta potential (). PCV-2 has highest quantity of DCP resulting in such values of zeta potential. PCV-3 gave lowest vesicle size and polydispersity index with moderate zeta potential and highest entrapment efficiency among the PC vesicles. PCV-3 had highest content of cholesterol which is reported to increase the entrapment efficiency. PC vesicles showed the drug release upto 14–20 h: PCV-1 showed upto 14 h, PCV-2 upto 16 h and PCV-3 upto 20 h (). PCV-3 contained highest amount of cholesterol which might be controlling the release of drug and also maintaining vesicle bilayer integrity. PCV-3 was selected for further study since it showed drug release upto 20 h ().
Figure 2. Cumulative drug release profile (%).
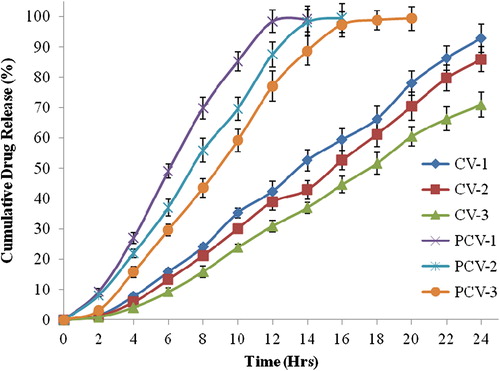
Table II. Physical characterization of formulations (PC vesicles and Ceramide vesicles).
In ceramide vesicle category, CV-1 showed the lowest vesicle size; however, the maximum vesicle size in ceramide vesicle category was 125 nm for CV-2, only 4 nm larger than minimum vesicle size. CV-1 has lowest quantity of the total lipid content and palmitic acid. All other components were equal so the variation in different physical parameters would be the result of variation in palmitic acid content and interaction of various lipid components in vesicle membrane. The lower vesicle size of ceramide vesicles could be explained by presence of positively charged lipid, ceramide-2 and negatively charged lipid cholesterol sulfate and palmitic acid in vesicle membrane, yielding tight packing. For a negatively charged drug, the electrostatic interactions could well be the reason of higher entrapment efficiency in ceramide vesicles. CV-1 showed the most negative zeta potential which helped in reducing aggregation of vesicles. On the other hand, lower values of negative charge in CV-3 resulted in higher drug entrapment. In ceramide vesicles, maximum entrapment efficiency was observed in CV-3. In vitro drug release study in ceramide vesicles was quite promising. All the ceramide formulations showed drug release upto 24 h. Maximum release was in CV-1, however, CV-3 was selected for further studies since it retained almost 29.04% drug content even after 24 h ().
Based on physical parameters and in vitro drug release, PCV-3 and CV-3 were selected for stability studies. The selected formulations were evaluated at three different temperature and humidity conditions (4 ± 0.5 °C, 25 ± 0.5 °C/60% RH and 40 ± 0.5 °C/75% RH) for 3 months.
Physicochemical parameters and in vitro drug release for PCV-3 after stability studies are shown in . It shows that at 4 ± 0.5°C, PCV-3 showed negligible change in vesicle size while increasing the temperature upto 40°C increases the vesicle size upto 167.2%. Polydispersity index was modified at increased temperature and humidity, whereas at 40 ± 0.5°C/75% RH, polydispersity index increased by 91.08%. The zeta potential was slightly affected by temperature and humidity modification. Entrapment efficiency was altered by temperature as storage for 3 months at 25 ± 0.5°C/60% RH and 40 ± 0.5°C/75% RH causes decrease by 53.77% and 83.71%, respectively. Most drastic effect was observed during in vitro release study of PCV-3. When freshly prepared, the vesicle formulation gave 99.4% drug release in 20 h which was retained during storage at 4 ± 0.5°C for 1 month. Further storage at 4 ± 0.5°C resulted in shortening in duration of release for maximum drug content. However, the sample stored at elevated temperatures and humidity conditions caused the release of 99.5% and 99.27% drug in 8 and 2 h, respectively, in 1 month. Storage at 25 ± 0.5°C/60% RH and 40 ± 0.5°C/75% RH any further did not give any release data because the formulation showed clumps.
Table III. Stability study of selected phosphatidylcholine vesicle (PCV-3).
Stability studies for CV-3 showed a different pattern as shown in . The vesicle size of the formulation did not vary much; however, storage at high humidity for 3 months (40 ± 0.5°C/75% RH) resulted in enlargement (5%) of vesicles. Similarly uniformity of size distribution was less in sample stored for 3 months at 40 ± 0.5°C/75% RH. Zeta potential of the formulation was unaffected at 4 ± 0.5°C. The samples stored at 25 ± 0.5°C/60% RH and 40 ± 0.5°C/75% RH after 3 months showed an increase in zeta potential which was to the tunes of 20%. The content of drug entrapped in CV-3 was 79.5% which was retained in samples stored at 4 ± 0.5°C and 25 ± 0.5°C/60% RH for complete duration of stability studies. The sample kept at 40 ± 0.5°C/75% RH showed the presence of appreciable content of drug entrapped after 1 month. After 3 months, total difference in entrapped drug content was 12.8%. Similar trend was observed for in vitro drug release in CV-3 formulation. CV-3 released 70.96% drug in 24 h. Storage at 4 ± 0.5°C, 25 ± 0.5°C/60% RH and 40 ± 0.5°C/75% RH increased the drug release by 1.95% and 2.9% and 12.5% in 3 months.
Table IV. Stability study of selected ceramide vesicle (CV-3).
Ceramide 2 is skin lipid component which retain the water content of skin to keep it elastic and hydrated. Its concentration varies with age leading to cutaneous desiccation and roughness. It may be possible that upon storage at such high humidity level, ceramide content absorb moisture and vesicles became swollen resulting in larger vesicles. Due to the hydration in vesicle membrane, the drug release was also facilitated despite the presence of intact vesicle membrane. This size increase earlier reflected in zeta potential which is a function of charge and surface area. Increased size of vesicle yielded decrease in surface area resulting in comparatively less negative zeta potential after 3 months storage.
The severe effect of temperature on phosphatidylcholine could be responsible for instability of vesicle membrane at elevated temperatures. At higher temperature, PC converts to lyso PC by means of hydrolysis through Deacylation/Reacylation process. Lyso PC has a less residue of fatty acid, thereby unable to support the vesicle membrane completely. Ceramide 2 used in CV vesicles has different phase behaviour and membrane properties at elevated temperature and it maintained stable vesicle membrane.
Based on the study, it could be inferred that phosphatidylcholine vesicles are most affected by temperature variations during storage and should be stored under strict temperature regulations. Further, the changes in PC membranes are irreversible and chemical in nature leading to vesicle rupture and leakage of drug content over a period of time. The vesicles containing ceramide-2 are improved in terms of stability owing to the less effect of temperature and humidity variations during typical storage conditions. Based on above study it can be assumed that PC liposomes should be stored at 4°C for not more than 1 month to keep them as effective as freshly prepared; however, ceramide vesicles can be stored at ambient room temperature for almost 3 months. This shows that ceramide vesicles are more stable vesicular formulation.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alam MN, Ahmad F J, Sultana Y, Iqbal Z, Aqil M, Ali A. 2008. Development and characterization of liposome based formulation of amiloride hydrochloride. J Dispersion Sci Technol. 29:415–420.
- An K, Sun Y, Xu L, Cui X. 2011. Preparation and in vitro evaluation of simvastatin ethosome. Artif Cells Blood Substit Immobil Biotechnol. 39:347–350.
- Arifin DR, Palmer AF. 2005. Stability of liposome encapsulated hemoglobin dispersions. Artif Cells Blood Substit Immobil Biotechnol. 33:113–136.
- Baillie A, Florence A, Hume L, Muirhead G, Rogerson A. 1985. Preparation and properties of niosomes-nonionic surfactant vesicles. J Pharm Pharmacol. 37:863–868.
- Baydoun L, Muller-Goymann CC. 2003. Influence of n-octenylsuccinate starch on in vitro permeation of sodium diclofenac across excised porcine cornea in comparison to Voltaren ophthalmic gel. Eur J Pharmaceut Biopharm. 56:73–79.
- Blume G, Cevc G. 1992. Drug-carrier and stability properties of the long-lived lipid vesicles, cryptosomes, in-vitro and in-vivo. J Liposome Research. 2:355–368.
- Dragicevic-Curic N, Scheglmann D, Albrecht V, Fahr A. 2008. Temoporfin-loaded invasomes: development, characterization and in vitro skin permeation studies. J Control Rel. 127:59–69.
- Gaur PK, Mishra S, Gupta VB, Rathod MS, Purohit S, Savla BA. 2010. Targeted drug delivery of rifampicin to the lungs: formulation, characterization, and stability studies of preformed aerosolized liposome and in situ formed aerosolized liposome. Drug Dev Ind Pharm. 36:638–646.
- Gaur PK, Mishra S, Kumar A, Panda BP. 2014a. Development and optimization of gastroretentive mucoadhesive microspheres of gabapentin by Box-Behnken design. Artif Cells Nanomed Biotechnol. 42:167–177.
- Gaur PK, Purohit S, Kumar Y, Mishra S, Bhandari A. 2014b. Preparation, characterization and permeation studies of a nanovesicular system containing diclofenac for transdermal delivery. Pharmaceut Dev Tech. 19:48–54.
- Goñi FM, Alonso A. 2006. Biophysics of sphingolipids I. Membrane properties of sphingosine, ceramides and other simple sphingolipids. Biochim Biophys Acta. 1758:1902–1921.
- Ishida T, Takanashi Y, Doi H, Yamamoto I, Kiwada H. 2002. Encapsulation of an antivasospastic drug, fasudil, into liposomes, and in vitro stability of the fasudil-loaded liposomes. Int J Pharm. 232:59–67.
- Kostarelos K. 2003. Rational design and engineering of delivery systems for therapeutics: biomedical exercises in colloid and surface science. Adv Colloid Interface Sci. 106:147–168.
- Li X, Ji J, Wang X, Wang Y, Shen J. 2007. Stability and drug loading of spontaneous vesicles of comb-Like PEG derivates. Macromolecul Rapid Comm. 28:660–665.
- Lintner K, Mondon P, Girard F, Gibaud C. 1997. The effect of a synthetic ceramide-2 on transepidermal water loss after stripping or sodium lauryl sulfate treatment: an in vivo study. Int J Cosmetic Sci. 19:15–26.
- Liu D, Jiang S, Shen H, Qin S, Liu J, Zhang Q. 2011. Diclofenac sodium-loaded solid lipid nanoparticles prepared by emulsion/solvent evaporation method. J Nanopart Res. 13:2375–2386.
- Moore DJ, Rerek ME. 2000. Insights into the molecular organization of lipids in the skin barrier from infrared spectroscopy studies of stratum corneum lipid models. Acta Dermatologica Venereol Supp. 208:16–22.
- Nasr M, Mansour S, Mortada ND, Elshamy AA. 2008. Vesicular aceclofenac systems: a comparative study between liposomes and niosomes. J Microencap. 25:499–512.
- Sinico C, Manconi M, Valenti M, Fadda AM. 2005. Liposomes as carriers for dermal delivery of tretinoin: in vitro evaluation of drug permeation and vesicle-skin interaction. J Control Rel. 103: 123–136.
- Touitou E, Dayan N, Bergelson L, Godin B, Eliaz M. 2000. Ethosomes – novel vesicular carriers for enhanced delivery: characterization and skin permeation properties. J Control Rel. 65:403–418.
- Walters KA, Roberts MS. 2002. The structure and function of skin. In: Walters KA, Ed. Transdermal and Dermatological Formulations. New York: Marcel Dekker INC, pp. 1–41.
- Wartewig S, Neubert RHH. 2007. Properties of ceramides and their impact on the stratum corneum structure: a review. Skin Pharmacol Physiol. 20:220–229.
- Weber TM, Kausch M, Rippke F, Schoelermann AM, Filbry AW. 2012. Treatment of xerosis with a topical formulation containing glyceryl glucoside, natural moisturizing factors, and ceramide. J Clin Aesthet Dermatol. 5:29–39.
- Wertz PW, Abraham W, Landmann L, Downing DT. 1986. Preparation of liposomes from stratum corneum lipids. J Inves Dermatol. 87:582–584.
- Yadav AV, Murthy MS, Shete AS, Sakhare S. 2011. Stability aspects of liposomes. Ind J Pharm Edu Res. 45:402–413.
- Yardley HL, Summerly R. 1981. Lipid composition and metabolism in normal and diseased epidermis. Pharmacol Ther. 13:357–383.
- Zheng S, Zheng Y, Beissinger RL, Fresco R. 1992. Liposome-encapsulated hemoglobin processing methods. Biomater Artif Cells Immobilization Biotechnol. 20:355–364.