
Abstract
Polymerized porcine hemoglobin (pPolyHb) exhibits a protective effect on ischemia/reperfusion of organ grafts. A series of experiments were performed to explore the underlying cytoprotective mechanisms of pPolyHb pretreatment on H2O2-induced cell death and apoptosis. The results showed that the pretreatment augmented heme oxygenase-1 (HO-1) expression, and at the same time, decreased the phosphorylation of JNK/p38 mitogen-activated protein kinase (MAPK) and intracellular ROS generation in H2O2-treated HUVECs. Moreover, the inhibition of HO-1 expression by tin porphyrin (SnPP) abolished the protective effects of pPolyHb, which suggested that the cytoprotective effect of pPolyHb involves upregulating HO-1 and subsequently decreasing the phosphorylation of the JNK and p38 MAPK and ROS generation.
Introduction
Ischemia/reperfusion (I/R) injury contributes to morbidity and mortality in a wide range of pathologies, including myocardial infarction, ischemic stroke, acute kidney injury, and trauma, and is still a major challenge during solid organ transplantation as well as cardiothoracic, vascular, and general surgery (CitationChang 2007). An imbalance in metabolic supply and demand within the ischemic organ results in profound tissue hypoxia and microvascular dysfunction. Therefore, many studies have focused on minimizing the adverse effects of I/R injury. Surgery, pharmacologic strategies, and gene therapy are the major methods currently used to alleviate I/R injury (CitationPapadopoulos et al. 2013).
Reactive oxygen species (ROS) are causal for I/R-associated tissue injury, which is a major contributor to organ dysfunction or failure (CitationAshraf et al. 2014). The first key step in the pathogenesis of I/R injury is ROS-induced vascular endothelial cell (EC) injury, since blood vessels are responsible for nutrient transfer and gas exchange. Attenuating oxidative stress-induced EC injury could reduce I/R injury in tissues. ROS are composed of several species, including hydrogen peroxide (H2O2), the hydroxyl radical (•OH), superoxide (O2•−), and singlet oxygen (•O). Among these, H2O2 is the dominant form in cells, as it is more stable than the other ROS (CitationRhee et al. 2003). Thus, H2O2 has been used extensively to mimic acute oxidative stress-induced EC injury.
Hemoglobin-based oxygen carriers (HBOCs) were initially designed as blood substitutes to efficiently deliver oxygen to hypoxic tissues in emergency situations. This ability suggests that HBOC may also be beneficial in alleviating I/R injury during the preservation of solid organs. It has been shown that preservation in the presence of HBOC during an ischemic period could attenuate I/R injury to the heart and kidney (CitationLi et al. 2012, CitationWu et al. 2011), and especially that pretreatment with HBOC is also able to provide a protective effect (CitationLi et al. 2011). Our previous studies have also illustrated that polymerized porcine hemoglobin (pPolyHb), a new HBOC, has potential protective effects on the preservation of the small bowel (CitationHuang et al. 2014). Additionally, Topp et al. have demonstrated that hemoglobin-glutamer (HbG)-200 can reduce cold preservation/reperfusion injury of rat liver (CitationTopp et al. 2008) by induction of heme oxygenase-1 (HO-1), although the underlying molecular mechanisms are not clearly understood.
The HO system is part of a vital cell signaling pathway that occurs in response to cellular injury or stress. The induction of HO-1 can catalyze heme to form CO, biliverdin/bilirubin (BV/BR), and Fe. The first two are thought to be effective in relieving oxidative stress. Over-expression of HO-1 may protect the liver from I/R injury by anti-inflammatory and anti-apoptotic effects (CitationParada et al. 2014). Some pharmacologic chemicals have been proven to protect against I/R injury by induction of HO-1, whereas HO-1 inhibition may abolish the protective effect and aggravate the damage (CitationMan et al. 2014, CitationXie et al. 2014). The protective effects of ischemic preconditioning and postconditioning on I/R injury have also been shown to be mediated by the induction of HO-1 (CitationWang et al. 2014). Since pPolyHb contains heme groups, we hypothesized that pretreatment with pPolyHb can protect against H2O2-induced injury by the induction of HO-1.
The mitogen-activated protein kinase (MAPK) family is one of the signaling pathways that have been implicated in the oxidative stress-induced cell death cascade (CitationRunchel et al. 2011). Among the MAPK family, the activation of the c-Jun N-terminal kinase (JNK)/p38 MAPK pathway is commonly linked to the promotion of cell apoptosis and death, particularly under conditions of oxidative stress (CitationAshraf et al. 2014, CitationKumphune et al. 2012), and thus JNK/p38 MAPK is also called stress-activated protein kinase (SAPK). Researchers have reported that in endothelial cells and cell lines, accumulation of H2O2 can activate the JNK/p38 MAPK pathway (CitationKumphune et al. 2012), whereas chemicals that inhibit the phosphorylation of JNK/p38 MAPK showed protective effects against H2O2-induced cell injury.
Therefore, in this study, we explored the potential effect of pretreatment with pPolyHb on H2O2-induced cell injury of HUVECs. We also investigated the underlying signal transduction mechanisms, with a focus on the induction of HO-1, and its influence on the JNK/p38 MAPK pathway.
Materials
Reagents
DMEM high glucose culture medium was purchased from HyClone (Logan, UT, USA). Fetal bovine serum was supplied by Gibco (Grand Island, NY, USA). 4′6-diamidino-2-phenylindole (DAPI), [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide] (MTT), and 29,79-dichlorofluorescein diacetate (DCFH-DA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-JNK, - p38 MAPK, -HO-1, -Bcl-2, -Bax, -cytochrome c, -caspase-3, -β-actin and -GAPDH antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The kits for the measurement of lactate dehydrogenase (LDH) concentrations were purchased from the Jiancheng Bioengineering Research Institute (Nanjing, Jiangsu, China). Anti-phospho-JNK (T183/Y185) MAPK and anti-phospho-p38 (T180/Y182) MAPK antibodies were purchased from ImmunoWay Biotechnology Company (Newark, DE, USA). The rabbit anti-goat, goat anti-rabbit, and goat anti-mouse secondary antibodies were purchased from Zhongshan Company (Beijing, China). Tin protoporphyrin IX (SnPP) (inhibitor of HO-1), SB203580 (inhibitor of p38 MAPK), and SP600125 (inhibitor of JNK) were all purchased from Sigma-Aldrich (St. Louis, MO, USA).
pPolyHb solutions
Glutaraldehyde-polymerized porcine hemoglobin (pPolyHb) was prepared as previously described (CitationZhu et al. 2011a, CitationZhu et al. 2011b). pPolyHb (10.5 ± 0.5 g/dL polymerized porcine hemoglobin, methemoglobin < 5%, endotoxin < 1.0 EU/mL, osmolality 300–330 mOsm, pH 7.4 ± 0.05, average molecular weight of pPolyHb 600 ± 50 kD, 64 kD tetramer < 2%) was formulated and stored at 4°C under nitrogen gas until use. Before being used, the pPolyHb was quickly mixed with phosphate-buffered saline solution (PBS) to the indicated final concentration, and then equilibrated with 95% O2 and 5% CO2 at 37°C for 30 min (pH 7.4) before use.
Methods
Cell culture
HUVECs (ATCC CRL-1730; Shanghai Tiancheng Technology Company, China) were cultured in DMEM high glucose medium supplemented with fetal calf serum (10%), 2 mM L-glutamine, 100 U/ml penicillin, and 100 g/ml streptomycin at 37°C in 5% CO2 and 95% air. After confluence, the cells were incubated in serum-free medium in the presence or absence of pPolyHb (5, 25, 125 μM) for 10 h, followed by exposure to 400 μM H2O2 for 4 h. The inhibitors were incubated with cells in serum-free medium for 2 h at 37°C and then removed before the treatments mentioned above. SnPP (20 μM) was added in advance of pPolyHb pretreatment, while SP600125 (20 μM) or SB203580 (20 μM) were added 2 h before exposure to 400 μM H2O2. The cells were harvested after the treatments, for further analysis.
MTT assay
Cell viability was measured using the MTT assay. Briefly, the cells were seeded into 96-well plates at a density of 1 × 104 cells/well. After incubation with different drugs for the indicated time periods, 10 μL of MTT solution (final concentration, 0.5 mg/ml) was added and incubation continued for 4 h at 37°C. The culture medium was then carefully removed, and 100 μL of DMSO was added to dissolve the formazan crystals, and the absorbance was measured using a microtiter plate reader (SpectraMax 190, Molecular Devices, USA) at a wavelength of 490 nm. Cell viability was expressed as an optical density (OD) value. In addition, cell morphology was observed using inverted/phase contrast microscopy, and images were obtained (Olympus BX61, Japan).
Lactate dehydrogenase (LDH) assay
LDH, an indicator of cell injury, was detected after exposure to H2O2, with an assay kit (Institute of Jiancheng Biotechnology, China), according to the manufacturer's protocol. Enzyme activity was expressed as units per liter, and the absorbance was measured at 440 nm. The results were expressed as percent of control groups.
Cellular apoptosis assay
Cellular apoptosis was analyzed by the DAPI assay using a cell death detection kit (Beyotime Institute of Biotechnology, China), according to the manufacturer's instructions. After the HUVECs were fixed in paraformaldehyde (4%) for 24 h, DAPI (0.5 mg/ml) was used (30 min at room temperature) to stain all the nuclei (blue), and the cells were observed using an 80i Fluorescence Microscope (Nikon Corporation, Tokyo, Japan). The apoptotic rate was expressed as the number of positively stained apoptotic HUVECs divided by the total number of HUVECs counted, multiplied by 100%.
Measurement of intracellular reactive oxygen species (ROS)
The measurement of intracellular ROS was based on the ROS-mediated conversion of nonfluorescent 29,79-DCFH-DA into fluorescent DCFH, as described previously (CitationRamalingam and Kim 2014). After the cells were seeded and treated in black 96-well plates, the cells were washed with PBS (pH 7.4), and then incubated with DCFH-DA (20 mM) in PBS at 37°C for 2 h. At the end of the incubation, the DCFH fluorescence of the cells in each well was measured at an emission wavelength of 530 nm and an excitation wavelength of 485 nm, using an FLX 800 microplate fluorescence reader (Biotech Instruments Inc., USA). The background condition was cell-free. The results were expressed as a percent of fluorescent intensity of the control group (100%). In addition, the cells were observed under FluoView FV1000 confocal microscopy (Olympus Corporation, Tokyo, JP), and images were obtained.
Western blot assay
After treatment with different protocols, the cells were washed and collected in ice-cold PBS and centrifuged (800 × g, 10 min). The cell pellet was resuspended in ice-cold lysis buffer containing 50 mmol/L Tris-HCl (pH 7.3), 150 mmol/L NaCl, 5 mmol/L EDTA, 1 mmol/L dithiothreitol, 1% Triton X-100, and 1% protease inhibitor cocktail, and incubated on ice for 30 min. The lysates were centrifuged (12,000 × g, 15 min), and the resulting supernatant was transferred to new tubes and stored at − 70°C. The protein concentrations were determined using the Bradford protein assay kit (Beyotime Institute of Biotechnology, China). The proteins were separated by SDS-PAGE electrophoresis and transferred to PVDF membranes. The membranes were blocked for 2 h in Tris-buffered saline and Tween 20 (TBST, pH 7.6) containing 5% non-fat dry milk powder, and thereafter incubated overnight at 4°C, with antibodies against HO-1, JNK, p38 MAPK, and the phospho forms of JNK and p38 (1:500 dilution), or Bcl-2, Bax, cytochrome c, caspase-3, GAPDH and β-actin (1:1000 dilution), followed by washing in TBST. The membranes were probed with different secondary antibodies (1:5000 dilution) at room temperature for 90 min, followed by washing in TBST. The protein bands were detected using chemiluminescence and quantified using the Quantity One software package (Bio-Rad Laboratories, UK). The results of the control group were defined as 100%.
Statistical analysis
Statistical analysis was performed using SPSS 11.5. Data were expressed as mean ± standard error of the mean for multiple comparisons. The data were analyzed by the one-way analysis of variance (ANOVA) followed by post-hoc Dunnett's t-test. P < 0.05 was considered statistically significant.
Results
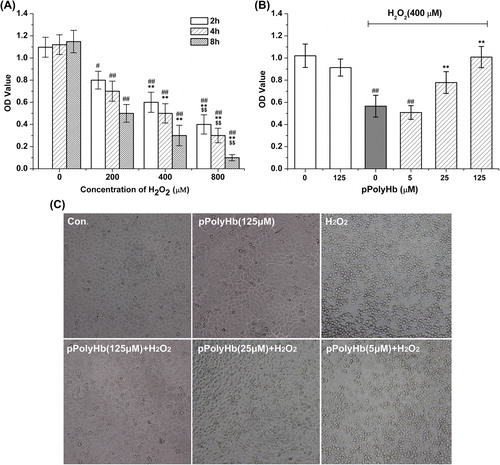
pPolyHb inhibits H2O2-induced HUVEC cell death
HUVECs were treated with H2O2 (200, 400, and 800 μM) for 2, 4, and 8 h. As expected, exposure to H2O2 at these concentrations significantly decreased the cell viability (P < 0.01 for all conditions, except P < 0.05 for exposure to 200 μM H2O2 for 2 h, compared to the respective control groups), and the cell viability was reduced by H2O2 in a concentration- and time-dependent manner (). The cell viability was reduced to nearly 46.3% of the respective control group after 4 h of exposure to 400 μM H2O2, and this condition was used to cause H2O2-induced cell injury in the subsequent experiments. Pretreatment with pPolyHb for 10 h was shown to improve the cell viability in a concentration-dependent manner (). In particular, pretreatment with 125 μM and 25 μM pPolyHb showed a significant increase in cell viability compared to that achieved with H2O2-treatment alone (P < 0.01). As observed using microscopy, the H2O2 treatment caused significant cell shrinkage and a decrease in the cellular attachment, compared to the control group. In contrast, pretreatment with pPolyHb increased the cell viability, attenuated H2O2-induced cell shrinkage, and improved the attachment of the cells (). Compared to the control group, treatment with pPolyHb alone had no effect on cell viability.
pPolyHb blocks H2O2-induced HUVEC cell apoptosis
The protective effects of pPolyHb on H2O2-induced apoptosis were determined. Pretreatment with pPolyHb inhibited H2O2-induced apoptosis in a concentration-dependent manner. As shown in , H2O2 induced obvious changes in the nuclear morphology of the HUVEC cell, such as heterogeneous intensity, chromatin condensation, and fragmentation, as observed using fluorescent microscopy. The percentage of apoptotic cells was 53.5 ± 4.5% (vs. control group, P < 0.01) in H2O2-treated cells. The apoptotic rate was significantly reduced to 35.3 ± 3.7% (P < 0.01) and 18.8 ± 4.0% (P < 0.01) in the 25 μM and 125 μM pPolyHb pretreatment groups, respectively. No difference in apoptosis was observed between groups treated with pPolyHb alone and the control group (P > 0.05).
Figure 2. Effect of pPolyHb on H2O2-induced apoptosis in HUVECs. (A) Cells were pretreated with the indicated concentrations of pPolyHb for 10 h, followed by 400 μM H2O2 (or no H2O2) for 4 h, and examined by morphologic analysis after DAPI staining. The arrowheads in the pictures indicate apoptotic cell nuclei (DAPI-positive cells).(B) Effects of pPolyHb on the expression of apoptosis-related proteins in H2O2-treated HUVECs. Cells were treated as in (A), then lysed, and Western blot analysis was performed. Bax, Bcl-2, caspase-3, and cytochrome c expression were analyzed. β-actin was used for normalization. Microscopy images and Western blot images are representative of three independent experiments. Data are means ± standard errors (n = 3). ##P < 0.01 compared to the control group; **P < 0.01 compared to the group treated with H2O2 alone.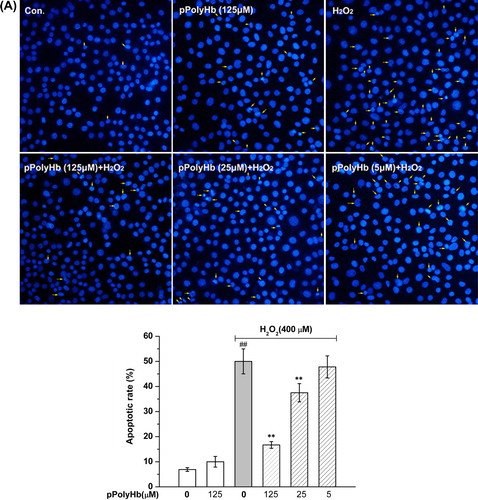
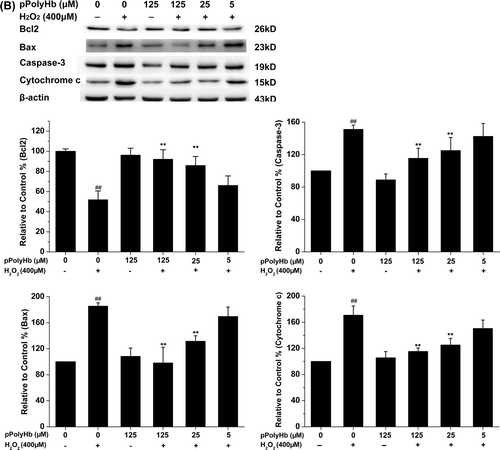
The effects of pPolyHb on the expression of the apoptotic regulatory proteins cytochrome c, caspase-3, Bax, and Bcl-2 were examined, to confirm its anti-apoptotic effect on H2O2-induced apoptosis. As shown in , H2O2 treatment resulted in a significant increase in cytochrome c, caspase-3, and Bax expression, and a decrease in Bcl-2 expression, compared to the control (P < 0.01). Pretreatment with pPolyHb significantly decreased caspase-3, cytochrome c, and Bax expression in H2O2-treated HUVECs, while increasing Bcl-2 expression (P < 0.01, compared to the H2O2-treated group) in a concentration-dependent manner. No differences were found between pPolyHb (125 μM) treatment alone and the control group in expression of these apoptotic regulatory proteins.
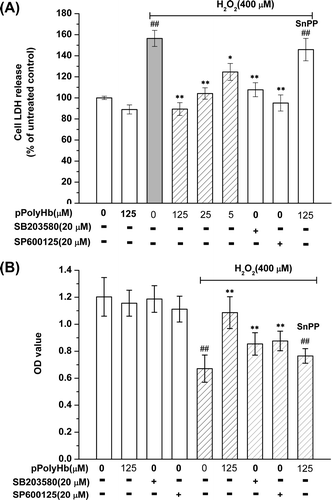
Effect of pPolyHb on H2O2-induced LDH leakage in HUVECs
LDH leakage from the cytoplasm into the culture medium, which reflects the integrity of the cell membranes, was determined under various conditions. As shown in , a significant increase in LDH leakage was found after 4 h of exposure to 400 μM H2O2, compared with the control group. Pretreatment with pPolyHb reduced LDH leakage in a concentration-dependent manner, which was consistent with the cell viability results (). Furthermore, to determine whether the induction of HO-1 and the JNK/p38 MAPK activation pathway play a role in H2O2-induced cell injury, LDH leakage and cell viability were examined using the inhibitors SnPP, SB203580, and SP600125. As shown in , pretreatment with SnPP greatly increased LDH leakage and decreased cell viability in the pPolyHb pretreatment groups, which suggested that HO-1 is involved in the cytoprotective effects of pPolyHb. Inhibition of JNK and p38 MAPK by their specific inhibitors significantly attenuated LDH leakage and increased cell viability during H2O2 treatment, compared to the results in the group treated with H2O2 alone ().
Pretreatment with pPolyHb induces HO-1 expression in HUVECs
The signal transduction pathway underlying the protective effect of pPolyHb against H2O2-induced cell injury was evaluated. Several studies have suggested that pPolyHb induces HO-1 expression and confers a protective effect against oxidative stress (CitationTopp et al. 2008). Therefore, we assessed the expression of HO-1 by the Western blot technique, following pPolyHb pretreatment. As shown in , pPolyHb pretreatment increased HO-1 expression in a concentration- and time-dependent fashion. After 10 h-incubation, 125 μM of pPolyHb induced a nearly 4-fold increase in HO-1 expression relative to the control group (), while the 50 μM and 25 μM concentrations also caused a significant increase in HO-1 expression. However, pretreatment with SnPP, the inhibitor of HO-1, abolished the pPolyHb-induction of HO-1 in HUVECs (). SnPP also abolished the protective effects of pPolyHb pretreatment on cell viability and LDH leakage following H2O2 treatment (). It is proposed, therefore, that pPolyHb exhibits its protective effect against H2O2-induced cell injury partly by upregulating HO-1.
Figure 4. pPolyHb induces HO-1 expression in a concentration- and time-dependent manner. (A) Cells were treated with 125 μM of pPolyHb for the indicated time (0, 2, 4, 6, 8, 10 h) and HO-1 expression was analyzed by Western blot. (B) Cells were treated for 10 h with the indicated concentrations (0, 125, 50, 25, 5 μM) of pPolyHb or pretreated with SnPP (20 μM) for 2 h, followed by treatment ith 125 μM pPolyHb, and then HO-1 expression was analyzed by Western blot. β-actin was used for normalization. **P < 0.01 compared to the control.
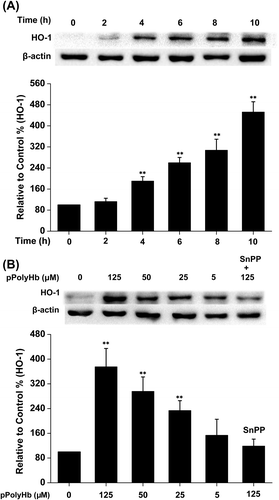
pPolyHb prevents H2O2-induced activation of JNK/p38 MAPK in HUVECs
MAPKs are involved in the regulation of apoptosis and anti-apoptosis, and the inhibition of JNK/p38 protected HUVECs against H2O2-induced cell injury (). Here, we investigated whether these two protein kinases play a role in the protective effect of pPolyHb during H2O2-induced apoptosis. As shown in , H2O2 increased the phosphorylation of the JNK54 isoform (JNK2) by 210% and the phosphorylation of the JNK46 isoform (JNK1) by 146% (P < 0.01). In addition, H2O2 increased the phosphorylation of p38 in HUVECs by 243% (P < 0.01). Compared to the results of treatment with H2O2 alone, pretreatment with pPolyHb reduced both the phosphorylation of JNK MAPK (JNK54 and JNK 46) and p38 MAPK at a concentration of 125 μM and 25 μM (P < 0.01) respectively. There was no significant difference in the measurement of pJNK (pJNK54 and pJNK46) or pp38 between the group treated with pPolyHb alone and the control.
Figure 5. Effects of pPolyHb on phosphorylation of JNK and p38 mitogen-activated protein kinase (MAPK) in H2O2-treated HUVECs. Cells were cultured in 6-well plates until confluent, and the medium was replaced with serum-free medium and cultured in the presence or absence of pPolyHb (5, 25, and 125 μM) for 10 h. Cells were then treated with 400 μM H2O2 for 4 h, followed by lysis and Western blot analysis. Densitometry scanning analysis of p-JNK and p-p38 MAPK phosphorylation was performed. GAPDH was used for normalization. Western blot images are representative of three independent experiments. Data are means ± standard errors (n = 3). ##P < 0.01 compared to the control, **P < 0.01 compared to the group treated with H2O2 alone.
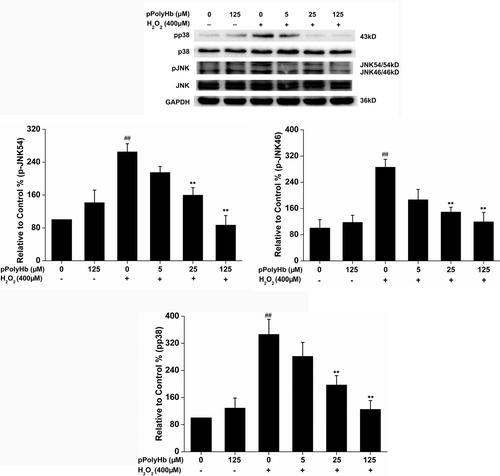
Effects of induction of HO-1 on the H2O2-induced phosphorylation of JNK and p38 MAPK
Since pretreatment of HUVECs with pPolyHb induces a significant increase in HO-1 () and inhibits the H2O2-induced phosphorylation of JNK and p38 MAPK (), the association of HO-1 induction with JNK and p38 MAPK phosphorylation was investigated. As shown in , H2O2 treatment alone caused a significant increase in phosphorylation of JNK54/46 and p38MAPK, whereas pretreatment with pPolyHb (125 μM) restored phosphorylation to the control level. Inhibiting pPolyHb induction of HO-1 by SnPP (20 μM) pretreatment, however, significantly increased the phosphorylation of JNK54/46 and p38 MAPK, compared to the pPolyHb pretreatment group (P < 0.01), to a level similar to that in the group treated with H2O2 alone. Taken together, our results elucidate the signal transduction pathway underlying the protective effects of pPolyHb against H2O2-induced cell injury in HUVECs: namely, that pretreatment with pPolyHb inhibits JNK and p38 MAPK protein kinase activation through the induction of HO-1.
Figure 6. SnPP attenuates the inhibitory effect of pPolyHb against H2O2-induced phosphorylation of JNK and p38 MAPK in HUVECs. Cells were treated with pPolyHb (125 μM), with or without pretreatment with SnPP (20 μM), followed by H2O2 (400 μM) incubation for 4 h. Western blot analysis was performed to determine the phosphorylation and protein expression of JNK and p38 MAPK. Normalization was performed with the anti-GAPDH antibody. Western blot images are representative of three independent experiments. ##P < 0.01 compared to control, **P < 0.01 compared to the group treated with H2O2 alone, $$P < 0.01 compared to the pPolyHb pretreatment group.
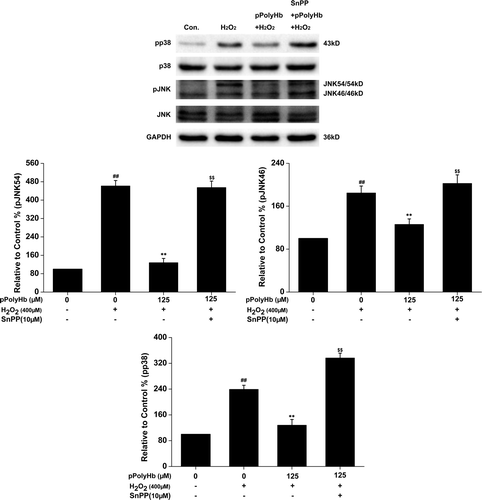
Inhibitory effect of pPolyHb on intracellular ROS generation in H2O2-treated HUVECs
Changes in ROS levels have been implicated in many aspects of HUVEC biology, including cell apoptosis and cytokine production. We determined whether pretreatment with pPolyHb elicited a potent ROS scavenging effect. shows representative confocal images of cells fluorescently stained with DCFH-DA. Using a fluorescence assay (), we found that HUVECs exposed to H2O2 showed increased intracellular ROS production (P < 0.01, 216.2 ± 10.8% vs control), whereas this increase was reduced to 189.1 ± 9.2%, 136.8 ± 8.6% (P < 0.01, vs H2O2-treated group), and 105.6 ± 8.9% (P < 0.01, vs H2O2-treated group) with pPolyHb pretreatment at doses of 5 μM, 25 μM, and 125 μM, respectively. However, treatment with pPolyHb alone showed no effects on intracellular ROS generation compared to the control. Pretreatment with the inhibitor of HO-1, SnPP, abolished the pPolyHb scavenging effect on ROS generation, suggesting that HO-1 is associated with the regulation of intracellular ROS generation.
Figure 7. Effects of pretreatment with pPolyHb on intracellular generation of reactive oxygen species (ROS). After 10 h of pretreatment with or without pPolyHb (5, 25, and 125 μM) or pretreatment with SnPP (10 μM) and pPolyHb (125 μM), HUVECs were exposed to H2O2 (400 μM) for 4 h. (A) Confocal microscopy images of cells fluorescently stained with DCFH-DA (magnification, × 40, scale bar = 50 μm). The microscopy images are representative of three independent experiments. (B) ROS generation was assayed using DCFH-DA fluorescence measured with a fluorometer (excitation = 485 nm, emission = 535 nm). Data are means ± standard errors (n = 3). ##P < 0.01 compared to the control; **P < 0.01 compared to the group treated with H2O2 alone.
Discussion
The vascular endothelium plays multiple roles in achieving adequate vascular tone and preventing platelet aggregation and microcirculatory dysfunction. I/R injury may lead to acute endothelial dysfunction and long-term graft vasculopathy in blood vessels (CitationGuibert et al. 2011). Therefore, the protection of endothelial function and cell viability is crucial in the preservation of grafts that contain abundant blood vessels. In the present report, H2O2 was used to mimic endogenous ROS generation, and the ability of pretreatment with pPolyHb, a novel HBOC, to protect against H2O2-induced HUVEC cell death and apoptosis, as well as the underlying mechanisms of action, were explored.
The majority drugs for anti-oxidative stress prevent apoptosis by regulating the intrinsic mitochondrial pathway (CitationChen et al. 2012). Bax, Bcl-2, cytochrome c, and caspase-3 play important roles in the oxidative stress-induced apoptotic process, and are all important members of the intrinsic mitochondrial pathway (CitationChen et al. 2012). No cytotoxic effects of pPolyHb were observed at 125 μM in the present culture system, and the addition of pPolyHb significantly protected HUVECs from H2O2-induced apoptosis. The balance between Bcl-2/Bax and the liberation of cytochrome c from mitochondria to the cytoplasm play crucial roles in apoptosis progression (CitationTzifi et al. 2012). In addition, cell apoptosis is mediated by an activation cascade of the caspase family of cysteine proteases that results in caspase-3 activation (CitationGu et al. 2014). Thus, apoptosis caused by H2O2 can reduce the Bcl-2/Bax ratio and increase the level of caspase-3 and cytochrome c in response to several factors such as oxidative stress and exposure to toxic substances. Our results showed that pPolyHb dose-dependently and significantly reduced the expression of the pro-apoptotic proteins Bax, caspase-3 and cytochrome c, while increasing expression of the anti-apoptotic protein Bcl-2 in H2O2-treated HUVECs (). In this regard, blocking of H2O2-induced apoptosis by pPolyHb appears to be mediated by the intrinsic mitochondrial pathway.
The expression of HO-1 (32–34 kD) is induced ubiquitously in response to oxidative stress, and confers cytoprotection in many models of lung and vascular injury and disease (CitationGozzelino et al. 2010). HO-1 induction also maintains tissue architecture, preserves organ function, and leads to prolonged graft survival (CitationLi et al. 2010). Vascular endothelial cells derived from HO-1−/− mice, or an HO-1-deficient child, exhibit enhanced susceptibility to oxidative stress in vitro (CitationYachie et al. 1999). The cytoprotective effects of HO-1 are related to end-product formation (CitationMorse et al. 2009), and the pharmacologic application of CO and BV/BR can mimic HO-1-dependent cytoprotection in many injury models. Hemoglobin-glutamer (HbG)-200 has been reported to exert a protective effect on cold hepatic preservation injury via upregulation of HO-1 (CitationTopp et al. 2008). Our study found an increase in HO-1 expression in HUVECs when co-incubated with pPolyHb, in a dose- and time-dependent manner. In particular, the cytoprotective effects against H2O2-induced HUVEC cell apoptosis increased at high concentration of pPolyHb, which corresponded to high HO-1-induction levels. This cytoprotective effect was subsequently attenuated by inhibition of HO-1 with SnPP in advance of pPolyHb pretreatment (), which indicates that pPolyHb reduces H2O2-induced oxidative stress injury by upregulation of HO-1.
JNK and p38 MAPK have been shown to be involved in antioxidant and anti-inflammatory responses, and are considered to be promising targets for the treatment of oxidative stress-induced injury. JNK and p38 MAPK are strongly activated by H2O2 and other chemical stressors, endotoxins, and inflammatory cytokines, especially in endothelial cells (CitationMorse et al. 2009). Although p38 MAPK was thought to be an upstream regulator of HO-1, some researchers have found that the anti-inflammatory (CitationKohmoto et al. 2007) and anti-apoptotic (CitationBrugger et al. 2010) effects of HO-1-derived CO are exerted via the p38 MAPK signal transduction pathway. Silva and colleagues have demonstrated that increased HO-1 activity degrades p38a in endothelial cells, suggesting that these two molecules might form a physiologically relevant feedback loop to regulate apoptosis (CitationSilva et al. 2006). Our study revealed that pPolyHb pretreatment reduced the phosphorylation of JNK and p38 MAPK in H2O2-treated HUVECs, which likely plays an important role in its ability to inhibit cell apoptosis. Moreover, the inhibition of induced HO-1 by SnPP abrogated both the cytoprotective effects of pPolyHb and its ability to inhibit the phosphorylation of JNK/p38 MAPK. Thus, we conclude that pPolyHb pretreatment inhibits JNK/p38 pathway-mediated apoptosis in H2O2-treated HUVECs via upregulation of HO-1.
Excessive intracellular ROS generation is known to be harmful to the endothelium of blood vessels, which may both directly and indirectly trigger cell death and cell apoptosis. The inhibition of HO-1 has been found to increase intracellular ROS levels and activate p38 MAPK in immature dendritic cells (CitationAl-Huseini et al. 2014). We observed an increase of intracellular ROS in H2O2-treated HUVECs, compared to the control group. However, pretreatment with pPolyHb dose-dependently suppressed ROS-generation in H2O2-treated HUVECs. This suppression could be abrogated by the inhibition of HO-1 production, suggesting that the induction of HO-1 plays a role in the decrease of intracellular ROS generation.
Conclusion
In summary, the present investigation suggests that pPolyHb pretreatment protects HUVECs against H2O2-induced cell death by increasing Bcl-2, and decreasing the expression of Bax, caspase-3 and cytochrome c. These effects are correlated with the upregulation of HO-1, inhibition of phosphorylation of JNK/p38 MAPK, and attenuation of intracellular ROS generation, which may all play important roles in mediating cell survival. Our results provide important insights into the protective mechanisms of pPolyHb in H2O2-induced cell death, and suggest that pPolyHb may have therapeutic potential to protect the vascular system from ischemia/reperfusion injury in applications such as cardiac surgery, organ preservation, and surgeries with extensive blood loss.
Acknowledgment
We would like to express our gratitude to Dr. Weixun Duan for his kindness in providing valuable suggestions for the experiments.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
We acknowledge with thanks the grants from National High-Tech R & D Program (863 Program) (Grant nos. 2012AA021902), National Natural Science Foundation of China (Grant nos. 81102367 and 31301405), China Scholarship Council of the Ministry of Education, as well as grants from the Shaanxi Science and Technology Department (Grant nos. 2011KTCL03-23, 2011K12-03-10 and 2012JM2009).
References
- Al-Huseini LM, Aw Yeang HX, Hamdam JM, Sethu S, Alhumeed N, Wong W, Sathish JG. 2014. Heme oxygenase-1 regulates dendritic cell function through modulation of p38 MAPK-CREB/ATF1 signaling. J Biol Chem. 289:16442–16451.
- Ashraf MI, Ebner M, Wallner C, Haller M, Khalid S, Schwelberger H, et al. 2014. A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury. Cell Commun Signal. 12:6.
- Brugger J, Schick MA, Brock RW, Baumann A, Muellenbach RM, Roewer N, Wunder C. (2010). Carbon monoxide has antioxidative properties in the liver involving p38 MAP kinase pathway in a murine model of systemic inflammation. Microcirculation. 17:504–513.
- Chang TMS. 2007. Artificial Cells: Biotechnology, Nanomedicinde, Regenerative Medicine, Blood Substiture, Bioencapsulation, and Cell/Stem Cell Therapy, Vol. 1 of Regerative Medicin, Artificial Cells and Nanomedicine. Singapore: World Scientific.
- Chen SD, Yin JH, Hwang CS, Tang CM, Yang DI. 2012. Anti-apoptotic and anti-oxidative mechanisms of minocycline against sphingomyelinase/ceramide neurotoxicity: implication in Alzheimer’s disease and cerebral ischemia. Free Radic Res 46:940–950.
- Gozzelino R, Jeney V, Soares MP. 2010. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50:323–354.
- Gu W, Zhang Q, Yin W, Li C. 2014. Caspase-3-mediated splenic lymphocyte apoptosis in a porcine model of cardiac arrest. Am J Emerg Med. 32:1027–1032.
- Guibert EE, Petrenko AY, Balaban CL, Somov AY, Rodriguez JV, Fuller BJ. 2011. Organ preservation: current concepts and new strategies for the next decade. Transfus Med Hemother. 38:125–142.
- Huang H, Ma J, Zhu W, Sun J, Yan K, Song B, et al. 2014. The application of polymerized porcine hemoglobin (pPolyHb) in the rat small bowel preservation. Artif Cells Nanomed Biotechnol. 42: 289–295.
- Kohmoto J, Nakao A, Stolz DB, Kaizu T, Tsung A, Ikeda A, et al. 2007. Carbon monoxide protects rat lung transplants from ischemia-reperfusion injury via a mechanism involving p38 MAPK pathway. Am J Transplant 7:2279–2290.
- Kumphune S, Chattipakorn S, Chattipakorn N. 2012. Role of p38 inhibition in cardiac ischemia/reperfusion injury. Eur J Clin Pharmacol. 68:513–524.
- Li LP, Zhang L, Peng LP, Cheng L. 2010. Heme oxygenase-1 is the candidate targeting for organ transplantation. Chin Med J (Engl). 123:2128–2134.
- Li T, Zhou R, Xiang X, Zhu D, Li Y, Liu J, et al. 2011. Polymerized human placenta hemoglobin given before ischemia protects rat heart from ischemia reperfusion injury. Artif Cells Blood Substit Immobil Biotechnol. 39:392–397.
- Li T, Zhang Z, Liao D, Chen Y, Yang C, Xu X, Liu J. 2012. The effect of polymerized placenta hemoglobin on renal ischemia/reperfusion injury. Artif Cells Blood Substit Immobil Biotechnol. 40:396–399.
- Man W, Ming D, Fang D, Chao L, Jing C. 2014. Dimethyl sulfoxide attenuates hydrogen peroxide-induced injury in cardiomyocytes via heme oxygenase-1. J Cell Biochem. 115:1159–1165.
- Morse D, Lin L, Choi AMK, Ryter SW. 2009. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radical Biol Med. 47:1–12.
- Papadopoulos D, Siempis T, Theodorakou E, Tsoulfas G. 2013. Hepatic ischemia and reperfusion injury and trauma: current concepts. Arch Trauma Res. 2:63–70.
- Parada E, Buendia I, Leon R, Negredo P, Romero A, Cuadrado A, et al. 2014. Neuroprotective effect of melatonin against ischemia is partially mediated by alpha-7 nicotinic receptor modulation and HO-1 overexpression. J Pineal Res. 56:204–212.
- Ramalingam M, Kim SJ. 2014. Insulin on hydrogen peroxide-induced oxidative stress involves ROS/Ca2 + and Akt/Bcl-2 signaling pathways. Free Radical Res. 48:347–356.
- Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. 2003. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 14:S211–215.
- Runchel C, Matsuzawa A, Ichijo H. 2011. Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Sign. 15:205–218.
- Silva G, Cunha A, Gregoire IP, Seldon MP, Soares MP. 2006. The antiapoptotic effect of heme oxygenase-1 in endothelial cells involves the degradation of p38 alpha MAPK isoform. J Immunol. 177:1894–1903.
- Topp S, Krieg A, Koch A, Tidden C, Ramp U, Hohlfeld T, et al. 2008. Hemoglobin-glutamer 200 reduces reperfusion injury of the cold preserved rat liver by induction of heme oxygenase-1. J Surg Res. 150:243–254.
- Tzifi F, Economopoulou C, Gourgiotis D, Ardavanis A, Papageorgiou S, Scorilas A. 2012. The role of BCL2 family of apoptosis regulator proteins in acute and chronic leukemias. Adv Hematol. 2012:524308.
- Wang Y, Shen J, Xiong XX, Xu YH, Zhang H, Huang CJ, et al. 2014. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. Plos One. 9:e98834.
- Wu W, Li T, Liu J, Yang C. 2011. Pretreatment before ischemia induction with polymerized human placenta hemoglobin (PolyPHb) attenuates ischemia/reperfusion injury-induced myocardial apoptosis. Artif Cells Blood Substit Immobil Biotechnol. 39:3–6.
- Xie Q, Li XX, Zhang P, Li JC, Cheng Y, Feng YL, et al. 2014. Hydrogen gas protects against serum and glucose deprivation-induced myocardial injury in H9c2 cells through activation of the NF-E2-related factor 2/heme oxygenase 1 signaling pathway. Mol Med Rep. 10:1143–1149.
- Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, et al. 1999. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 103:129–135.
- Zhu H, Dang X, Yan K, Dai P, Luo C, Ma J, et al. 2011a. Pharmacodynamic study of polymerized porcine hemoglobin (pPolyHb) in a rat model of exchange transfusion. Artif Cells Blood Substit Immobil Biotechnol. 39:119–126.
- Zhu H, Yan K, Dang X, Huang H, Chen E, Chen B, et al. 2011b. Immune safety evaluation of polymerized porcine hemoglobin (pPolyHb): a potential red blood cell substitute. Artif Cells Blood Substit Immobil Biotechnol. 39:398–405.