
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective
Ibuprofen is an established non-steroidal anti-inflammatory drug commonly used for general inflammation. However, it causes gastrointestinal troubles when administered orally, thereby decreasing patient compliance. Transdermal application of vesicular formulation of Ibuprofen will provide better patient compliance and efficacy.
Methods
Six different compositions of lipid constituents have been formulated into nanovesicles using thin-film hydration method and dispersed into gel using Carbopol 934. The formulations were characterized based on physicochemical parameters using photon correlation spectroscopy, transmission electron microscopy, in vitro drug release, ex vivo skin permeation using human skin, and in vivo studies.
Results
The formulation, ibuprofen liposomal gel-5 (ILG-5), had nanoliposome of smallest size (159 nm) and polydispersity index (0.331). This formulation showed moderate zeta potential and the highest encapsulation. All the formulations including IG showed a considerable amount of drug release through in vitro synthetic membrane. ILG-5 showed maximum permeation during skin permeation studies. IG showed no permeation in ex vivo settings. ILG-5 has shown the highest Cmax and AUC during in vivo permeation study.
Conclusions
The present work clearly shows the superiority of nanoliposome formulation over non-vesicular formulations and that lipid composition containing 7/3/1 molar ratio of phosphatidylcholine, cholesterol, and dicetyl phosphate is optimum for nanoliposome preparations, in the cases where controlled delivery of drug is needed for a sufficient period of time.
Introduction
Ibuprofen is a widely used anti-inflammatory drug; however, it can cause local mucosal irritations due to its acidic nature. Transdermal dosage forms are desirable for the chronic use of this drug, especially in the case of rheumatic symptoms. The efficacy of transdermal formulations depends greatly on its capacity to facilitate the drug penetration through the skin. In the present study, vesicular formulations were developed, optimized, and evaluated various parameters along with in vitro and skin permeation. CitationBula and Ghaly (1995) first reported the liposome of ibuprofen and evaluated the effects by varying the ratio of lipid to drug, the filter size, and the stirring period during hydration of the dried lipid layer. CitationNewa et al. (2007) prepared binary solid dispersion of ibuprofen using Poloxamer 188 and evaluated in vitro drug release and in vivo studies after oral administration. CitationBorovac et al. (2006) prepared ibuprofen-loaded polyvinyl alcohol-based hydrogel beads to alleviate side effects, such as inflammation and pain, following uterine artery embolization for the treatment of leiomyomata. Ntawukulilyayo prepared xanthan gum– n-octenylsuccinate starch matrix tablet containing ibuprofen and evaluated in vitro and in vivo drug release after oral administration (CitationNtawukulilyayo et al. 1996). CitationAl-Saidan (2004) observed an important phenomenon of transdermal self-permeation enhancement of ibuprofen and reported the permeation enhancer behavior of ibuprofen for itself and for diclofenac sodium. Rasool developed and evaluated chitosan gels of ibuprofen using several penetration enhancers, for example, propylene glycol, polyethylene glycol 400, oleic acid, glycerol, and menthol (CitationRasool et al. 2010). CitationShumilov et al. (2010) developed transdermal gel of ibuprofen and evaluated its pharmacokinetics and activity. CitationStephen et al. (2013) added peripheral vasodilators, tolazoline or papaverine, for increasing transdermal permeation of ibuprofen.
Nanoliposome, which is submicron bilayer lipid vesicle, is a new technique for encapsulation and drug delivery. Several diverse categories of compounds, that is, drugs, cosmetics, and nutraceuticals, can be incorporated into nanoliposomes. The applications of nanoliposomes are immense as they are biocompatible, biodegradable, and have size in nanometer range. It includes diagnosis, cancer therapeutics, gene delivery, cosmetics, food technology, and agriculture. Nanoliposomes can enhance the activity of bioactive agents by improving their solubility, bioavailability, and stability, and preventing unnecessary interactions with other molecules (CitationMozafari 2010).
CitationShuibing et al. (2013) characterized nanoliposomes for entrapping medium-chain fatty acids and vitamin C by lyophilization. CitationGye et al. (2013) prepared chitosan-coated nanoliposomes for improving the mucoadhesive property of curcumin using the ethanol injection method.
CitationJun-Bom et al. (2012) formulated nanoliposome for transdermal delivery of hydrophilic drugs. CitationGaur et al. (2014b) developed and evaluated nanovesicles for effective transdermal delivery. CitationGaur et al. (2014c) developed and evaluated permeation studies of a nanovesicular system containing diclofenac for transdermal delivery.
The objective of the present work was to devise nanoliposome formulations for non-steroidal anti-inflammatory drug (NSAID), ibuprofen. The developed formulation was meant for effective transdermal delivery. The formulations were evaluated for physicochemical parameters; in vitro drug permeation evaluated by means of an artificial membrane, ex-vivo permeation, in vivo study, and anti-inflammatory study. The formulation was judged against a gel formulation containing the free drug, ibuprofen. This work will prove to be useful for enhancement of permeation level of an effective and commercially used ibuprofen.
Materials and methods
Ibuprofen and phosphatidylcholine (PC) were gift samples from Asoj Soft Caps, Baroda, India. Cholesterol (Chol), synthetic cellulose acetate membrane (MWCO 12000 Da), and dicetyl phosphate (DCP) were purchased from Himedia, Mumbai, India. Solvents and other chemicals were of analytical grade and were obtained from commercial source.
Nanoliposome preparation
The nanoliposome was prepared by lipid film hydration method (). The constitutive lipids were dissolved in chloroform solution (20 ml) of drug in the appropriate ratio. The lipid–drug mixture was deposited as a thin film in a round-bottom flask by evaporating the solution under vacuum. Vacuum was applied for 1 h to ensure the total removal of traces of solvent. The film was hydrated at a temperature above the gel–liquid transition temperature with phosphate buffer saline (PBS). The resulting hydrated lipid–drug mixture was initially stirred at 10,000 rpm for 10 min (mechanical agitation; Remi, New Delhi, India) and then sonicated using a probe sonicator for 15 min. The obtained formulation was filtered through a membrane (Immobilon-P Membrane, 0.45 μm pore size; Millipore Pvt. Ltd., New Delhi, India). Unentrapped drug was removed by passing the formulation through Sephadex G-20 minicolumn (CitationTouitou et al. 2000, CitationGaur et al. 2014a, Citation2014b).
Table I. Composition of formulations (Drug lipid ratio = 1:3).
Preparation of gel
Gels were prepared by dispersing 1% w/w of Carbopol 934 in distilled water. These gels were stirred using mechanical stirrer. The dispersion was then neutralized (pH, 7.4) by addition of 1% w/w triethanolamine. Any entrapped air in the gel was allowed to escape by allowing the gels to stand overnight. The drug was added at concentration of 1% w/w by means of gentle levigation (CitationGaur et al. 2014a, Citation2014b).
Characterization of formulations
Photon correlation spectroscopy (PCS)
Determination of polydispersity index (PDI) and zeta potential was performed using Zetasizer Nano ZS, equipped with a 4-mW He–Ne laser (633 nm). The particle diameter range and number of photon counts per second (kilo counts per second) were evaluated at room temperature. The formulations were taken in lyophilized form in micro-centrifuge tubes, suspended in phosphate buffer at pH 7.4 and introduced into the instrument to read the results for determining PDI. For determination of zeta potential, formulations were taken in lyophilized form in 2-ml eppendrof tube, suspended in phosphate buffer at pH 7.4. All measurements were performed in triplicate for each sample.
Transmission electron microscopy
Transmission electron microscopy along with negative staining was used to determine the size and shape of nanovesicle. A drop of formulation dispersion was placed over a 200-mesh copper grid and allowed to absorb, and surplus was removed by filter paper. Then, phosphotungstic acid (1%) was added and surplus was removed by filter paper. The sample was dried at room temperature before imaging the formulations with Philips CM-10 (acceleration voltage: 100 kV; magnification: upto 4 50 000x; cryo-attachment).
Assay
Spectrophotometric analysis was used to determine the drug concentration. First, 25–500 μL of stock solution (10 mg/10 mL, adjusted with phosphate buffer at pH 7.4) was transferred into 10-mL volumetric flasks using microsyringe. Then, the volume was adjusted with phosphate buffer at pH 7.4. The absorbances of the solutions were determined against a blank spectrophotometrically (Shimadzu UV- 160A) at 277 nm. The aliquots of PBS without the drug were used as a blank. A calibration curve was then obtained (Y = 34.1X–0.603, in which Y = concentration [μg/mL] and X = absorbance). The sensitivity was 2.5–50 μg/mL. The limit of detection was 7.5 μg/mL.
Entrapment efficiency
The entrapment efficiency was determined using ultracentrifugation. Formulations were spun in a TL-100 ultracentrifuge (Beckman, USA) in a TLA-45 rotor overnight at 4°C, removed, and drug quantity was determined in both the sediment and the supernatant, following the procedure mentioned in assay. The entrapment efficiency was calculated as follows: [(T−C)/T] × 100, where T is the total amount of drug that is detected in both the supernatant and the sediment, and C is the amount of drug detected only in the supernatant (CitationTouitou et al. 2000, CitationGaur et al. 2014c).
Viscosity
The viscosity of the formulations was determined using Brookfield DV III ultra V6.0 RV cone and plate rheometer (Brookfield Engineering Laboratories, Inc., Middleboro, MA) using spindle # CPE40 at 25 ± 0.5°C (CitationAkhter et al. 2008).
In vitro drug release studies
Before the experiment, membrane (Himedia, Mumbai, India) was allowed to equilibrate at 37°C in physiological saline solution. It was placed over flow-through-type Franz diffusion cells. The acceptor compartment was filled with PBS (pH 7.4). The nominal surface area of the cell was 1 cm2. After equilibration, a finite dose of formulation was applied on the donor surface and the experiment was carried out under non-occlusion and light protection. Aliquots of 1 ml were taken out after every 2-h interval up to 24 h and the volume was replaced with fresh solution. Aliquots were analyzed for drug content using assay (CitationSinico et al. 2005, CitationGaur et al. 2014b).
Skin permeation studies (ex vivo studies)
The experiment was done as per the method described by CitationCuric et al. (2008) with suitable modifications. Full-thickness human skin obtained from plastic surgery patients was used for the permeation experiment. Subcutaneous fatty tissue was removed using a scalpel, and the surface of the skin was cleaned with the Ringer's solution. After drying, it was packed in aluminum foil and stored in a polyethylene bag at − 20°C.
Before the experiment, skin was allowed to thaw (37°C), cleaned with Ringer's solution, and transferred onto the flow-through-type Franz diffusion cells. The acceptor compartment was filled with PBS (pH 7.4), and epidermal side of the skin was exposed to ambient conditions for equilibration and kept overnight. The nominal surface area of the Franz cell was 1 cm2.
After equilibration, a finite dose of formulations (IG and ILG formulation containing 50 mg of ibuprofen) was applied on the skin surface, and the experiments were carried out under non-occlusion and light protection (CitationCuric et al. 2008, CitationGaur et al. 2014a). Aliquots of 1 ml were taken out after every 2-h interval up to 24 h. Aliquots were analyzed for drug content using the assay mentioned above.
In vivo skin permeation
The study was performed using 18 male albino rats. The animals were housed under standard laboratory conditions (temperature: 25 ± 2°C; relative humidity: 55 ± 5%) with free access to diet. The study was carried out using animals with an average weight of 260 g. For the experiment, the animals were anesthetized and their abdominal hairs were removed (CitationGaur et al. 2014a). The application site (3.14 cm2) was cleaned with distilled water. The treatments given were as follows:
Blood samples were removed at pre-decided time intervals and stored in vacutainer tubes. Plasma was separated by centrifugation at 5000 rpm for 20 min, and stored at − 21°C until drug analysis. The drug content was estimated using optimized high-performance liquid chromatography (HPLC) assay.
The instrument possesses a Shimadzu LC-10AT VP pump, a SIL-10AF auto injector, an SPD-10A UV–VIS detector, and an SCL-10A VP system controller HPLC system (Shimadzu, Japan). The method was as per CitationGaur et al. (2014a).
The column was Shim-pack VP-ODS, 4.6 mm I.D. × 150 mm, 5 μm column (Shimadzu, Japan), eluted with acetonitrile/0.1 M sodium acetate/acetic acid (70:30:0.5, v/v/v) isocratically at a flow rate of 2.0 ml/min. The injection volumes for standard and plasma samples were 20 μL. The detector was set at 223 nm for ibuprofen detection.
Anti-inflammatory study
Carrageenan-induced rat paw edema model using Wistar albino rats was used for determination of anti-inflammatory activity. Eighteen rats (260 g, three groups) were kept under standard laboratory conditions (details in in vivo study). IG and ILG-5 were applied on the skin (3.14 cm2) in Groups I and II, respectively. Standard drug—ibuprofen (IOS) was administered orally at a dose of 10 mg/Kg in Group III. The untreated paw was considered as negative control. The treatment was applied before using carrageenan. The edema was determined using digital plethysmometer (water displacement method) at 0, 1, 2, 3, 4, 5, and 6 h.
Edema inhibition (%) was estimated to establish the anti-inflammatory activity as per the following formula (CitationGaur et al. 2014a):
Skin irritation studies in animals
A skin irritation test was conducted on both sexes of rats to determine the potential irritation response based on the previously described method (CitationMichael et al. 2005). The groups of rats were divided into three groups. Group I was treated with saline, Group II with drug-loaded nanoliposome, and Group III with 5% solution of sodium lauryl sulfate. The formulation was held at the application site using a bandage for 24 h. After that, the dressing and patch were removed and the skin residue was cleaned with water. Skin irritation effects were assessed at approximately 0, 24, 48, and 72 h after the removal of the dressings. The adjacent areas of untreated skin from each animal served as control. Erythema and edema were scored on a scale of 0–4, with 0 showing no effect and 4 representing severe erythema or edema. The specific degree of irritancy was obtained by calculating the Primary Dermal Irritation Index (PDII) and classified according to the descriptive rating for mean PDII (slight irritation: 0 ≤ PDII ≤ 2.0; moderate irritation: 2.0 ≤ PDII ≤ 5.0; severe irritation: PDII ≥ 5.0).
Data and statistical analysis
The cumulative amount of drug released through a unit area of skin was plotted as a function of time. Steady-state flux (Jss) and lag time (tlag) were obtained from slope and x-intercept of the linear portion, respectively. Permeability coefficient (Kp) and diffusion parameter (D) were calculated from the following equations:
where D is diffusion coefficient within the skin (cm2 h− 1), h is diffusional path length, and Cd is the initial drug concentration in donor compartment.
Data were expressed as mean of 3 values ± S.D (standard deviation) except for permeation studies, where mean of 6 values ± S.D was used for calculations. Statistical analyses were performed using the PCP Disso software. Statistical comparisons were made using analysis of variance and statistical significance was set at P < 0.05.
Results and discussion
Results
All the formulations showed formation of circular vesicles which were in the size range of 159–174 nm (). The PDI were in the range of 0.331–0.461. All the formulations showed negative zeta potential as a result of incorporation of DCP ().
Figure 1. Transmission electron micrograph of ILG-5.
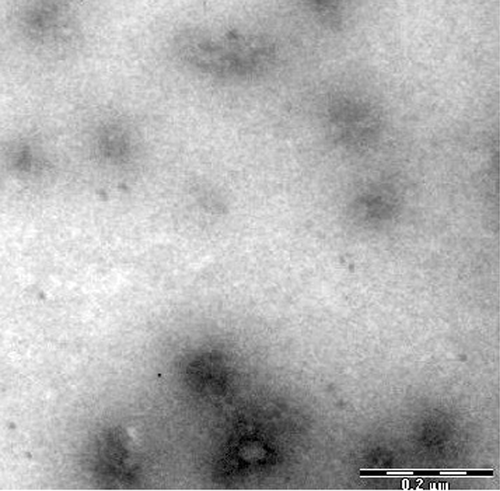
Table II. Physicochemical parameters of various formulations.
Maximum entrapment efficiency was displayed by ILG-5 at 49%, whereas all other formulations showed entrapment efficiency within the range of 33–49%. Upon gel formation, all the formulations have shown similar viscosity ().
Carbopol gel containing free drug has shown the highest drug release through artificial membrane, whereas vesicle formulations have shown drug release from 81 to 92% over a period of 24 h ().
Figure 2. Pattern of in vitro drug release in 24 h for different formulations.
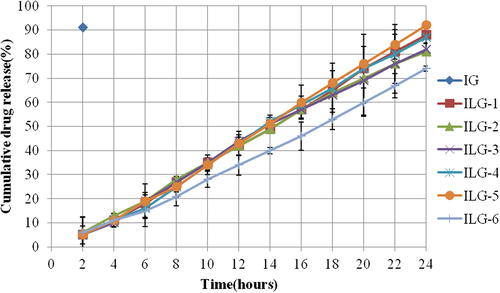
IG containing free drug has shown burst release since the interval between two data points was 2 h in our study. At the same time interval (2 h), maximum drug release from the vesicular formulation was 6%, showing the controlled release nature of vesicular formulations.
IG did not show any drug permeation in ex vivo settings, while vesicular formulations showed that 11–15% of drug content permeated (). The steady-state drug flux, lag time, permeability coefficient, and distribution parameter were calculated on the basis of ex vivo drug permeation data (). Maximum drug flux was shown by ILG-5 at 0.635, which also showed minimum lag time of 0.613.
Figure 3. Pattern of ex vivo drug permeation in 24 h for different formulations.
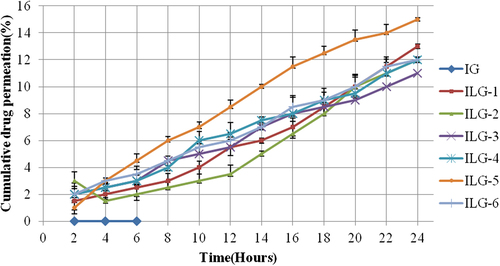
Table III. Different permeation parameters for nanoliposome formulations.
On the basis of ex vivo drug permeation, ILG-5 was selected for in vivo evaluation (). Cmax values were 0.68, 2.8, and 7.35 μg and tmax values were 6, 2, and 6 h for IG, IOS, and ILG-5, respectively. The AUC values for ILG-5 were 14.88 and 5.51 times the AUC values of IG and IOS, respectively, at 110.1 μg.h/ml ().
Figure 4. In vivo drug permeation study.
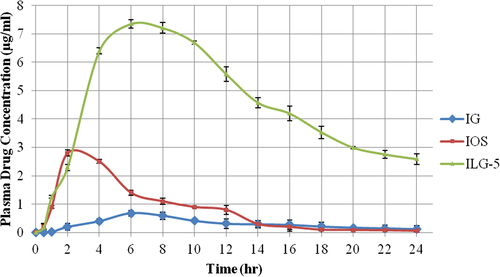
Table IV. In vivo study data of the IG, IOS, and ILG-5.
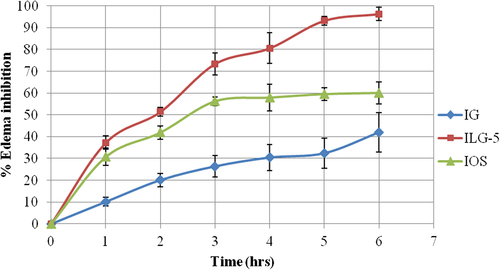
ILG-5 was compared with IG and a standard anti- inflammatory treatment (IOS, peroral) using carrageenan-induced rat paw edema model for determination of anti-inflammatory activity. Maximum protection against carrageenan insult was provided by ILG-5, which has shown 37.23% edema inhibition in initial hours of edema production (). ILG-5 has shown 1.21 times higher edema inhibition in comparison with standard drug—ibuprofen—in initial hours and 3.67 times higher edema inhibition in comparison with IG formulation.
Figure 5. Anti-inflammatory activity.
All the groups treated with Ibuprofen gel have shown minor edema after 3–4 days. No group in nanoliposome gel caused any erythema or dermal reactions, and nanoliposome gel formulations have irritation index of not more than 0.5.
Discussion
Transdermal drug delivery is an important mode of delivery of drugs, but an appropriate type of formulation should be selected for using this to deliver hydrophilic drugs. Liposome is an important category of lipidic formulations which can be used for increasing transdermal permeation of hydrophilic drugs, such as ibuprofen.
The total lipid content increases as we progress with the vesicular formulation series. The vesicular formulations contain DCP as charge-bearing moiety. Usually, it resides in vesicle walls and imparts it a net negative charge, as is evident from negative zeta potential. Another implication of this electrostatic charge could be attraction or repulsion based on the type of electrostatic charge on the drug molecule. Ibuprofen bears a negative charge in ionic state as it is an acid derivative, but it is located inside the core of vesicle. Location of drug molecules will also play a critical role in determining vesicle size. On the other hand, lipid composition and mixing speed are the determining factors for vesicle size which results in lower values of PDI.
As discussed earlier, the content of DCP imparts a net negative charge which plays an important role in reducing adhesion and fusion of nanoliposome; however, it also makes them susceptible toward human immune response. Zeta potential is affected by magnitude of charge and surface area of the nanoliposome. Furthermore, it is affected by spatial localization of various components in the nanovesicle wall.
In vitro drug release using synthetic membrane indicates the efficiency of formulation for drug release. The developed vesicular formulations were compared with a gel-containing free drug to understand the effect of vesicular membrane on drug delivery. All the vesicular formulations have shown to control drug release. PC and Chol are the main lipid components and are the determinants of vesicle membrane nature and permeability. PC has a phase transition temperature at 41°C which would result in existence of gel-state nanoliposome at physiological temperature, whereas Chol acts as membrane stabilizer by increasing permeability and decreasing order in gel-state nanoliposome. The effect of PC and Chol was evident in in vitro drug release. The results of ex vivo drug permeation can be corroborated by the presence of Chol and sphingolipids in skin. Chol increases permeability of PC vesicles, whereas sphingolipids present in upper layers of skin interact with nanoliposome and get inserted in place of Chol. These hybrid nanoliposomes can work as the reservoir of drug and mainly govern the drug release from nanoliposome.
ILG-5 showed similar results in in vivo study and anti-inflammatory study. Carrageenan-induced edema has two phases, first phase occurs in 1–2 h and second phase in 3–5 h. In the second phase, IL-1 activates polymorphic nuclear cells for producing lysosomal enzymes and active oxygen, resulting in connective tissue damage and paw swelling. Rat skin is thinner than human skin so edema inhibition was more pronounced in activity determination, since corresponding ex vivo study was done on human skin.
Conclusion
Nanoliposomes are an important carrier for delivering drug to the dermal compartment and for transdermal delivery; however, the selection of appropriate lipid phase holds the key to a successful vesicular carrier. From the present study, it is concluded that the lipid phase containing PC, Chol, and DCP in molar ratio of 7/3/1 is the optimum for ibuprofen nanoliposome formulation for efficient transdermal delivery.
Acknowledgements
The authors acknowledge Asoj Soft caps for providing ibuprofen and phosphatidylcholine.
Further, we acknowledge the sophisticated analytical instrumentation facility, AIIMS, New Delhi for TEM analysis and Advanced centre for nanotechnology, National Institute of Pharmaceutical Education & Research for providing facility of Zetasizer nano ZS.
Declaration of interest
The authors report no declarations of interests. The authors solely are responsible for the content and writing of the paper.
References
- Akhter S, Jain GK, Ahmad FJ, Khar RK, Jain N, Khan IZ, Talegaonkar S. 2008. Investigation of nanoemulsion system for transdermal delivery of domperidone: Ex-vivo and in vivo studies. Curr Nanosci. 4:381–390.
- Al-Saidan SM. 2004. Transdermal self-permeation enhancement of ibuprofen. J Control Release. 100:199–209.
- Borovac T, Pelage JP, Kasselouri A, Prognon P, Guiffant G, Laurent A. 2006. Release of ibuprofen from beads for embolization: In vitro and in vivo studies. J Control Release. 115:266–274.
- Bula D, Ghaly ES. 1995. Liposome delivery systems containing ibuprofen. Drug Dev Ind Pharm. 21:1621–1629.
- Curic ND, Scheglmann D, Albrecht V, Fahr A. 2008. Temoporfin-loaded invasomes: Development, characterization and in vitro skin penetration studies. J Control Release. 127:59–69.
- Gaur PK, Mishra S, Purohit S, Kumar Y, Bhandari A. 2014a. Development of a new nanovesicle formulation as transdermal carrier: Formulation, physicochemical characterization, permeation studies and anti-inflammatory activity. Artif Cells Nanomed Biotechnol. 1–8: In-Press.
- Gaur PK, Purohit S, Kumar Y, Mishra S, Bhandari A. 2014b. Ceramide-2 nanovesicles for effective transdermal delivery: development, characterization and pharmacokinetic evaluation. Drug Dev Ind Pharm. 40:568–576.
- Gaur PK, Purohit S, Mishra S, Kumar Y, Bhandari A. 2014c. Preparation, characterization and permeation studies of a nanovesicular system containing diclofenac for transdermal delivery. Pharm Dev Technol. 19:48–54.
- Gye HS, Seoung KC, Jun TK, Hee JJ, Hyun JP. 2013. Preparation of chitosan-coated nanoliposomes for improving the mucoadhesive property of curcumin using the ethanol injection method. J Agric Food Chem. 61:11119–11126.
- Jun-Bom P, Hyung-gon N, Jae-Hyuk J, Jung-Mi K, Chin-Yang K. .(2012). Enhanced transdermal delivery and optimization of nano-liposome preparation using hydrophilic drug. J Pharm Investig. 42:57–63
- Michael S, Taharat Y, Anthony EK, Aimee LL, Jason B, Karrie AB. 2005. Safety and toxicological evaluation of a novel niacin bound chromium (III) complex. J Inorg Biochem. 99:2161–2183.
- Mozafari MR. 2010. Nanoliposomes: preparation and analysis. Methods Mol Biol. 605:29–50.
- Newa M, Bhandari KH, Li DX, Kwon TH, Kim JA, Yoo BK, et al. 2007. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int J Pharm. 343: 228–237.
- Ntawukulilyayo JD, Vervaet C, Remon JP, Gortz JP, Berlo JA. 1996. In vitro and in vivo evaluation of a xanthan gum-n-octenylsuccinate starch matrix tablet containing ibuprofen as a model drug. Int J Pharm. 139:79–85.
- Rasool BKA, Abu-Gharbieh EF, Fahmy SA, Saad HS, Khan SA. 2010. Development and evaluation of ibuprofen transdermal gel formulations. Trop J Pharm Res. 9:355–363.
- Shuibing Y, Chengmei L, Wei L, Haixia Y, Huijuan Z, Wei Z, Yaqin H. 2013. Preparation and characterization of nanoliposomes entrapping medium-chain fatty acids and vitamin C by lyophilization Int J Mol Sci. 14:19763–19773.
- Shumilov M, Bercovich R, Duchi S, Ainbinder D, Touitou E. 2010. Ibuprofen transdermal ethosomal gel: characterization and efficiency in animal models. J Biomed Nanotechnol. 6:569–576.
- Sinico C, Manconi M, Peppi M, Lai F, Valenti D, Fadda AM. 2005. Liposomes as carriers for dermal delivery of tretinoin: in vitro evaluation of drug permeation and vesicle-skin interaction. J Control Release. 103:123–136.
- Stephen GC, Zhen Z, Gyula V, Aristidis V, Jim ER. 2013. Vasomodulation influences on the transdermal delivery of ibuprofen. J Pharm Sci. 102:4072–4078.
- Touitou E, Dayan N, Bergelson L, Godin B, Eliaz M. 2000. Ethosomes – novel vesicular carriers for enhanced delivery: characterization and skin penetration properties. J Control Release. 65:403–418.