
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Aim of the study was to develop solid lipid nanoparticles (SLN) of triamcinolone acetonide (TA) and to study the effect of various process variables in order to optimize the formulation for effective delivery. Drug loaded SLNs were successfully prepared and characterized by TEM, XRD and DSC study. Process variables like surfactant concentration, drug concentration, lipid concentration etc. showed significant effect on the particle size and entrapment efficiency. SLNs exhibited prolonged drug release following Higuchi release kinetics (R2 = 0.9909). In vitro skin distribution study demonstrated systemic escape of drug from TA loaded SLNs which might eliminate side effects associated with systemic exposure.
Introduction
Corticosteroids are commonly used to treat dermal disorders like psoriasis, atopic dermatitis, vitilgo etc. (CitationPradhan et al. 2013). But many available conventional therapies with corticosteroids are either cosmetically unacceptable, inconvenient for long-term use, or associated with dose dependant side effects. Moreover poor aqueous solubility of corticosteroids has limited their use in dermatological application. For all these reasons, compliance with treatment is reduced, and the lack of effective and safe treatment for these dermal disorders has created need to develop and implement novel approach to overcome these problems with a view to make the therapy more useful and acceptable.
Novel approach may include the use of some colloidal drug carriers (e.g. micelles, nanoemulsions, nanosuspensions, liposomes, polymeric nanoparticles) but they possess several drawbacks like limited physical stability, aggregation, drug leakage on storage, presence of solvent residues left over from production and cytotoxicity related problems with the use of polymers (CitationSingh et al. 2011, Citation2012, Citation2013, Citation2014). Hence, the research has been diverted towards the use of biocompatible lipids as carriers for the delivery of poorly soluble drugs to reduce the above stated problems (CitationPouton 2006). Solid lipid nanoparticles (SLNs) have emerged as a potential delivery carrier to resolve this problem.
SLNs are colloidal carrier systems composed of high melting point solid lipid core, coated by surfactants (CitationGhadiri et al. 2012). SLNs offer distinct advantages like negligible skin irritation, controlled release and protection of substances as it uses physiologically tolerated, non-irritative and non-toxic lipids (CitationProw et al. 2011). They are also safe options for use on inflamed and damaged skin. Moreover, small size of the lipid particles ensures its close contact with stratum corneum thereby increasing the amount of the drug penetrating into the mucosa or skin. These nanosized particles with their solid lipid matrix may also permit for sustained release of drugs (CitationSchafer-Korting et al. 2007). Occlusive properties of SLNs have been reported after topical application, which decreases transepidermal water loss and favours the drug penetration through the stratum corneum (CitationProw et al. 2011). They have also been shown to improve the dermal localization of several topical therapeutic agents (CitationChen et al. 2006, CitationLopes et al. 2006).
Triamcinolone acetonide (TA) is a glucocorticoid used in management of skin disorders like psoriasis. Their clinical effectiveness in the treatment of psoriasis is related to their vasoconstrictive, anti-inflammatory, immunosuppressive, and antiproliferative effects (CitationLiu et al. 2008).
Currently, researchers have been focusing mainly on the development of strategies to improve the benefit-risk ratio of glucocorticosteroids. SLNs as a delivery carrier could be a better option with a view to improve aqueous solubility and therapeutic concentration of triamcinolone acetonide in the target tissue, allowing long-term release of the drug at the target site thereby reducing the dose dependant side effects. Therefore, exploring the potential of SLNs loaded with TA appears valuable.
The present work deals with the optimization of various formulation parameters, such as homogenization time (HT), sonication time (ST), surfactant concentration (SC), lipid concentration (LC), drug concentration (DC) and homogenizer speed (HS) for the formulation of TA loaded SLNs by emulsification–ultrasonication method. Solubility of the drug in the solid lipids was determined to screen the suitability of lipid for formulation. The effects of the process variables on the particle size, poly dispersity index (PDI), zeta potential (ZP), drug entrapment efficiency (EE) and drug loading (L) of SLNs were investigated. The SLNs were subjected to physicochemical characterization by transmission electron microscopy, powder X-ray diffraction (PXRD) and Differential scanning calorimetry (DSC). Drug release study and drug distribution study through skin were also performed to observe the drug release profile from the developed SLNs and to evaluate the potential of drug loaded SLNs to deliver drug in epidermal and dermal region via an in vitro method. Finally, stability of the developed formulation was evaluated.
Materials and methods
Materials
TA was a generous gift from Ciron drugs Pvt Ltd (India). Compritol® 888 ATO, Precirol® ATO5 was a kind gift from Gattefossé (France). The dialysis membrane (Molecular weight cut off 10,000–12,000 Da) was purchased from HiMedia Laboratories (India). Methanol, soya lecithin, poloxamer, glycerol monostearate and stearic acid were purchased from Sigma Aldrich (India). Purified water from ultra-pure water system (Synergy UV water purifier system, India) was used throughout the study.
Measurement of drug solubility in lipids
Due to solid nature of the lipids, equilibrium solubility study was not possible; therefore, an alternative method was used in order to measure solubility of drug in the solid lipids (CitationDas et al. 2011). Solubility of TA was tested against four lipids Compritol® 888ATO, Precirol® ATO5, glycerol monostearate and stearic acid. In brief, 10 mg TA was accurately weighed and placed in a screw capped glass bottle covered with aluminium foil. Approximately, 200 mg of lipid was added into the bottle and heated at 80°C with continuous stirring. Further, extra amount of lipid was added in portions with constant stirring and heating at 80°C until a clear solution was obtained. Total amount of lipid added to obtain a clear solution was recorded.
Formulation procedure
SLNs were prepared by modified emulsification– ultrasonication method (CitationDas et al. 2011). Briefly, the lipid phase containing lipid, drug and acetone was heated to 70°C (temperature above melting point of the lipid), an aqueous solution of poloxamer 188 and soya lecithin at the same temperature was added and the pre-emulsion was stirred with UltraTurrax T10 (T-10 basic IKA) at 10,000 rpm for 5 min. The pre-emulsion was then ultrasonicated for 10 min under heating (Frontline sonicator, India) followed by pouring into cold (1–4°C) water and stirred with a magnetic stirrer. The SLNs were allowed to recrystallize at room temperature. Formulations of SLNs were prepared by varying enlisted variables as per design given in , in which components used are as follows:
Table I. Formulation design for SLNs.
Mean particle size and polydispersity index
The mean particle size and PDI of TA loaded SLNs were determined by Malvern Zetasizer ZS 90 (Malvern Instruments Inc., UK). Samples were prepared by dispersing SLN in sufficient amount of ultra-purified water prior to the experiment.
Zeta potential measurement
Zeta potential (ZP) reflects the electric charge on the particle surface. It is a useful parameter to predict the physical stability of colloidal systems (Patel et al. 2014). ZP was determined by Malvern Zetasizer Nano ZS (Malvern Instruments Inc., UK). Calculation of zeta potential is carried out using the Helmoltz–Smoluchowski equation:
Where, ζ is zeta potential, EM is electrophoretic mobility, η is viscosity of the dispersion medium and ϵ is the dielectric constant. Prior to the measurement, all samples were diluted using ultra-purified water.
Percent entrapment efficiency and drug loading
The bound drug (entrapped) in the SLNs dispersion was sedimented by controlled centrifugation at 12,000 rpm, 4°C for 15 min and the supernatant was decanted without disturbing the SLNs pellet. Fixed volumes of supernatant were diluted in methanol and analysed for unentrapped drug using UV spectrophotometer at 239 nm (PerkinElmer, Lambda 25, UK). % EE and % L was calculated using the following formula.
Transmission electron microscopy
The morphological examination of TA loaded SLNs was performed with Transmission Electron Microscopy (model TECNAI 200 KV TEM-Fei, Electron Optics, Japan). A drop of diluted sample was placed on the surface of the carbon coated copper grid and stained with negative stain using a drop of 2% (w/w) aqueous solution of phosphotungstic acid for 30 s. Excess staining solution was washed out by filter paper, leaving a thin aqueous film on the surface. After staining, samples were dried at room temperature for 10 min to carry out investigation (CitationRawat et al. 2007).
X-ray diffraction study
XRD study was performed to analyse crystalline or amorphous nature of the TA loaded SLNs. X-ray powder diffraction studies of pure TA, Compritol® 888ATO, physical mixture of TA and Compritol® 888ATO (at a ratio equivalent to that in the formulation) and lyophilized TA loaded SLNs were carried out using powder X-ray diffractometer (PANalytical 3 kW X'pert Powder, UK). Samples were placed on the sample stage and scanned from 2° to 60° with an operating voltage of 40 kV and current 30 mA.
Differential scanning calorimetry study
DSC studies of pure TA, Compritol 888ATO, physical mixture of TA and Compritol 888ATO (at a ratio equivalent to that in the formulation) and the lyophilized SLNs powder were carried out. Accurately weighed samples were carefully placed in DSC boats and heating curves were recorded in the temperature range of 25–350°C at a heating rate of 10°C/min under a constant flow of dry nitrogen. The study was carried out using Differential Scanning Calorimeter (Shimadzu DSC-60 Systems, Japan) equipped with a computerized data station TA-50WS/PC.
In vitro release study
In vitro release studies of TA loaded SLNs and plain TA suspension was carried out using a modified Franz diffusion cell as reported by CitationKhurana et al. (2013a). A dialysis membrane with molecular weight cut-off of 10,000–12,000 Da was used for the study. Prior to mounting in the Franz diffusion cell, the membrane was soaked in double-distilled water for about 12 h. Phosphate buffer with pH 7.4 was used as the diffusion medium. Whole assembly was put on a magnetic stirrer at 300 rpm and the temperature was maintained at 37.0 ± 0.5°C. The samples (plain TA suspension and TA loaded SLNs dispersion) equivalent to 2 mg drug were kept over the membrane in the donor compartment and stirred. Samples were withdrawn from the receptor compartment at predetermined time intervals (0, 1, 2, 3, 4, 5, 6, 8, 10, 12 and 24 h) and the volume was refilled with the diffusion medium of the same volume. The samples were analysed spectrophotometrically at 239 nm after appropriate dilutions. The % drug release was calculated and graph of % drug release against time was plotted, release studies were performed in triplicate for each formulation.
In vitro skin distribution study
In vitro skin distribution studies of TA loaded SLNs and plain TA suspension was carried out using goat skin. Samples of goat skin were obtained from the local slaughter house (Raipur, Chhattisgarh) to evaluate the in vitro skin distribution of TA delivered via TA loaded SLNs suspension and plain TA suspension. The permeation studies were performed using the Franz diffusion cells described above according to previously described methods with slight modification (CitationGupta and Trivedi 2014, CitationKhurana et al. 2013a). Treated skin samples with formulation containing drug equivalent to 400 μg (6 h with each test formulation) were processed at the conclusion of the experiment. The skins were washed and removed from the Franz cells. Epidermal and dermal layers were manually separated using tweezers. TA was extracted in 5 ml methanol with homogenization of each skin layer, and extracted samples were assayed for TA concentration by UV spectroscopy at 239 nm.
Stability studies
The stability of the developed SLN formulation was evaluated for 3 months according to CitationDas et al. (2011). Briefly, samples were stored in the sealed amber coloured glass vials at 4°C. After 3 months, the samples were characterized with respect to the particle size, ZP, PI, EE and L.
Results and discussion
TA loaded SLN were prepared by the modified emulsification–ultrasonication method varying various parameters as given in . These parameters were evaluated in terms of the particle size, ZP, PI, EE and L. Based on the evaluation parameters, the final batch with desirable properties was prepared (selected batch).
Solubility of TA in lipids
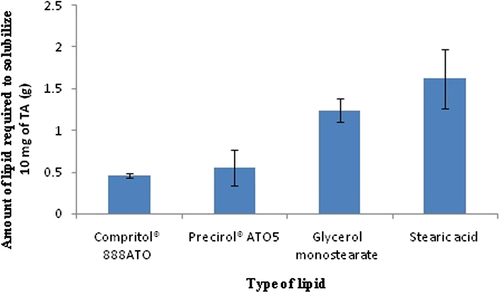
Solubility of a drug in lipid is one of the most important factors for determining drug loading capacity of the SLNs (CitationCharoenputtakhun et al. 2014). Results from the solubility studies are shown in . Solubility of TA was tested in four lipids: Compritol® 888ATO, Precirol®ATO5, glycerol monostearate and stearic acid. Among them, Compritol® 888ATO showed highest solubilization capacity followed by Precirol® ATO5 with least solubility in stearic acid. Amount of glycerol monostearate (1.24 ± 0.14 g) and stearic acid (1.62 ± 0.36 g) required to solubilize 10 mg TA was significantly higher than Precirol® ATO5 (0.55 ± 0.21 g) and Compritol® 888ATO (0.46 ± 0.03 g). This study indicated that TA loading capacity of Precirol® ATO5 and Compritol® 888ATO might be more than glycerol monostearate and stearic acid.
Figure 1. Solubility study of drug (Triamcinolone acetonide) in various lipids.
Effects of process variables
The modified emulsification–ultrasonication method was found to be efficient and quick to produce SLNs. Effect of different process variables on size, PDI, ZP, EE and L are discussed in the following sections and presented in .
Table II. Size, PDI, ZP, EE, and L of the drug loaded solid lipid nanoparticles (data represent mean ± SD).
Homogenization time
HT did not show any remarkable effect on the particle size and can be correlated to the fact that the homogenization step is only for emulsification of lipid in the aqueous phase. Further it does not produce final particles; rather, it is an intermediate step. Generally, ZP value indicates the degree of repulsion between close and similarly charged particles in the nanodispersion and predicts physical stability of the nanodispersion (CitationLason et al. 2013). High ZP (negative or positive) indicates stable nanoparticle dispersion as highly charged particles are responsible for prevention of aggregation of the particles due to electric repulsion. In this case, highest ZP (−34.2 ± 0.11 mV) was observed in the formulation with 5 min homogenization time. EE was slightly lower in case of 1 min HT, and maximum EE was observed with 10 min HT, which might be due to poor dispersion of nanoparticles in 1 min as compared to better dispersion of nanoparticles at 10 min HT. However, with 10 min HT the highest PDI and particle size was achieved which may be due to development of charge on the surface of nanoparticles leading to aggregation of smaller nanoparticles into larger sized particles. Based on the results of the particle size, PDI, zeta potential and % L, 5 min homogenization time was selected as optimum HT for TA loaded SLN formulation.
Surfactant concentration
Concentration of the surfactant demonstrated its huge influence on the particle size and PDI. The particle size decreased with increasing surfactant concentration (SC) from 0.5% to 4% w/v. This might be due to better stabilization of internal structure of dispersion at higher surfactant concentration preventing coalescence. PDI decreased with increasing SC up to 1%w/v. These observations might be due to production and stabilization of smaller lipid droplets (nanoemulsion) at higher SC as enough surfactant was present to stabilize the nanodroplets. However, in this case PDI was higher surprisingly at 2% w/v SC and 4% w/v SC. According to the literature published, the nanoparticles having PDI lower than 0.25 are considered relatively homogeneous, with minimum tendency for aggregation (CitationMitri et al. 2011). SC at 2% w/v and 4%w/v showed ZP value of − 29.9 ± 0.91 mV and − 29.2 ± 0.8 mV, respectively, which predicted poor physical stability of the SLN dispersions. In our observation, EE and L increased with increase in concentration of SC. This could be due to the presence of sufficient SC which helped the drug to remain within the lipid particles and/or on the surface of the particles. Low concentration of TA in SLN and high compatibility between the drug and the lipid further contribute to high EE. CitationLiu et al. (2007) also reported similar results for isotretinoin-loaded solid lipid nanoparticles. They demonstrated decrease in the particle size with significant increase in % EE with increase in the concentration of surfactant. Based on the results of investigation 1% w/v SC, which produced particles with optimum particle size, PDI, zeta potential, EE and % L was chosen as the optimum SC required for preparation of TA loaded nanoparticles.
Lipid concentration
The particle size significantly increased with increasing lipid concentration (LC). This may be correlated to better distribution of sonication energy in the dilute (low LC) dispersion as compared to the concentrated dispersion, which was responsible for more efficient particle size reduction at lower LC. Numerous studies have reported that increasing lipid content results in larger particles and broader particle size distribution (CitationTrotta et al. 2001, CitationSubedi et al. 2009). In this case, the particle size was lowest in case of 1% w/v LC with very high PDI (0.515) and highest at 10% w/v LC with 0.316 PDI. In all LCs, ZP was more than − 28 mV. As expected, EE increased with increasing LC. This might be due to availability of higher amount of lipid for drug encapsulation at high LC which led to higher EE. Opposite tendency was noticed for low LC. In contrary, % L significantly decreased with increasing LC. DC remained same in spite of increased LC, which led to reduction of drug to lipid ratio (i.e., loading) with increasing LC. This observation was in accordance with CitationHao et al. (2012). They developed baicalin loaded solid lipid nanoparticles and reported positive influence of high lipid concentrations on EE. By this observation, 2% w/v LC was selected to achieve best results because with 1%w/v LC a poorly stable system with high PDI was observed whereas at higher LC of 5%w/v and 10%w/v very large sized particles with lower % L were observed. However, with 2%w/v optimum particle size, PDI, zeta potential, higher EE and % L were observed.
Sonication time
Sonication time (ST) showed marked influence on the particle size and PDI. The particle size and PDI significantly decreased with increasing ST. These observations are reasonable as sonication was responsible for the final particle size of SLNs, which breaks the coarse emulsion drops to nanoemulsion droplets. Longer sonication time puts more sonication energy to the SLN dispersions, which reduced size of the nanoemulsion droplets and decreased size distribution. This observation was in accordance with Vitorino et al. They prepared SLN of simvastatin and reported that sonication reduced both the particle size and PDI (CitationVitorino et al. 2011). In the present study, we observed ZPs to be − 28.1 ± 0.81 mV and − 27.4 ± 0.8 mV for 10 min and 20 min ST, respectively, but for 10 min sonication time ZP was − 34.2 ± 0.19 mV indicating a stable system at 10 min sonication time. ST of 5 min produced particles with comparatively lowest EE. Otherwise, EE and % L were not significantly different among the groups. Samein also reported less impact of sonication time on EE of nystatin loaded SLN prepared using glyceryl monostearate as the lipid constituent (CitationSamein et al. 2014). Based on the observation of the particle size and stability aspect, 10 min ST was chosen as the optimum value for preparation of TA loaded nanoparticles as with ST less than 10 min (5 min ST) very large particle size was observed and with ST more than 10 min (15 min and 20 min ST) poor stability of formulation was observed.
Drug concentration
There were remarkable differences in particle size among different batches prepared with drug concentrations (DC) from 0.025%w/v to 0.2%w/v. PDI also increased with higher DC. This observation can be correlated to the presence of high amount of drug. ZPs were observed to be more than − 33.1 ± 0.1 mV in all cases, which suggests good stability of the SLN dispersions. EE decreased with increasing DC whereas % L increased with increase in DC upto 0.1%w/v. EE directly depends on the amount of drug added pertaining to the encapsulation capacity of the lipid used (CitationPatil et al. 2014). As the lipid has a certain drug loading capacity, addition of excess drug led to increase of the unencapsulated drug (i.e. decrease of EE). Similarly, % L cannot increase beyond maximum drug loading capacity signifying that loading has reached its maximum concentration of 0.1% w/v DC. But at 0.1% w/v DC the highest particle size with high PDI was observed indicating a poor stability of formulation.
Therefore, on the basis of the particle size, PDI, EE and % L, DC with 0.05% w/v was selected for the preparation of the final drug loaded SLN formulation. Similar observation was observed by CitationDas et al. (2012).
Homogenizer speed
The final size of the nanoparticles in the process depends on the globule size throughout the emulsification process. In this study, increase in the homogenizer speed led to decrease in the particle size but above 10,000 rpm there was no significant reduction in the mean particle size. Moreover, effect of the homogenizer speed was found insignificant in case of EE and L. Similar result was reported by Mulla and Khaji who developed GMS nanoparticles using soy lecithin as stabilizer at a constant concentration of 5.0% (w/w) and fixed homogenization time of 10 min. They reported a decrease of the nanoparticle mean size with an increase of the homogenizer speed. But above 13,500 rpm, there was no significant reduction of the particle size (Mulla and Kazi 2012). In our study, 10,000 rpm was selected for further preparation of drug loaded SLN. Therefore, the final formulation was prepared using 0.05% w/v DC, 2% w/v LC, 1% w/v SC, 5 min HT, 10 min ST and 10,000 rpm HS as final values of variables.
Mean particle size and polydispersity index
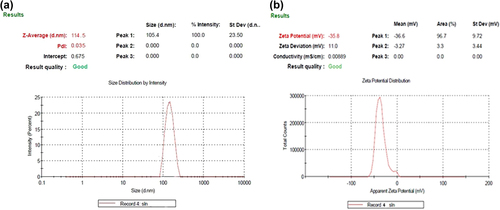
The mean particle size of TA loaded SLNs were found to be in the range of 94.01 ± 1.92 nm to 388.41 ± 1.13 nm with PDI of 0.045 to 0.316. However, the mean particle size for the selected batch was obtained to be 114.5 ± 1.32 nm with PDI of 0.035 ().
Figure 2. (a) Mean particle size and polydispersity index of selected batch formulation; (b) Zeta potential of the formulation of the selected batch.
Zeta potential
The zeta potential of drug loaded SLNs were found to be in the range of 27.4 ± 0.8 mV to 42.2 ± 0.13 mV. The zeta potential of the selected batch was obtained to be − 35.8 ± 0.61 mV (). The zeta potential value of the selected batch indicated the stable formulation.
Percent entrapment efficiency and loading
% EE of the prepared drug loaded SLN was found to be in the range of 30.19 ± 1.18 to 89.12 ± 2.22. For the selected batch % EE and % L was obtained to be 82.0 ± 1.32 and 1.71 ± 0.032. High entrapment can be attributed to the lipophilic nature of the drug having higher affinity for the selected lipid matrix.
Transmission electron microscopy
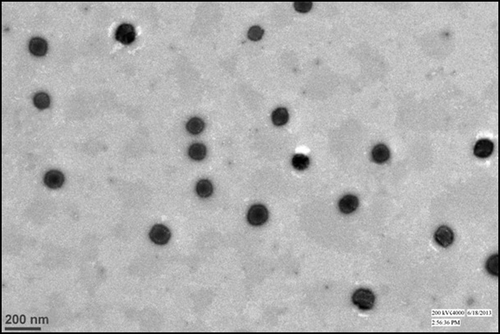
Shape and size of the selected batch of nanoparticles were evaluated by TEM. TEM images of the SLNs confirmed that the particles were nearly spherical or oval in shape with narrow size distribution and were non-aggregated (). The diameters of the particles observed in the micrographs are in good agreement with the data obtained from the Malvern particle size analyzer.
Figure 3. TEM image of the selected batch of TA loaded solid lipid nanoparticle.
X-ray diffraction study
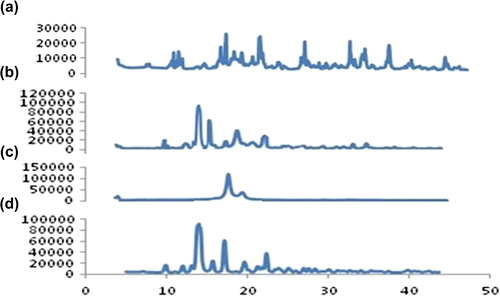
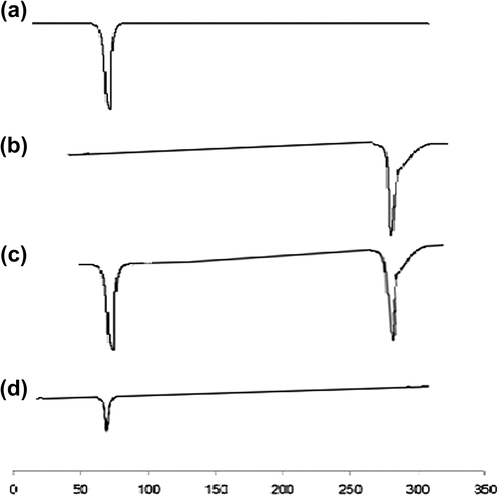
A sharp diffraction peak appeared in spectra of pure TA at 9.89, 12.0, 14.01, 15.7, 17.1, 19.6, 23.3 and 25.1 (). Compritol ATO 888 showed a characteristic peak at 2° value of 4.3, 21.3 and 23.5 (). In the physical mixture, the crystalline peak for TA was clearly evident (), whereas the SLN formulation showed a deformed peak for TA, indicating the relative reduction in the diffraction intensities in the TA loaded SLN. This could be further predicted due to the change in orientation of crystals or reduction in the quality of crystals of TA, and this change in diffraction pattern supports conversion of the crystalline drug to an amorphous form and contributing in the enhancement of solubility of the drug ().
Figure 4. XRD analysis. (a) Pure TA (b) Compritol® 888ATO (c) Physical mixture of TA and Compritol® 888ATO (d) TA loaded SLNs.
Differential scanning calorimetry
DSC is a tool to investigate the melting and recrystallization behaviour of crystalline material like SLNs (CitationAgrawal et al. 2010). shows the DSC thermograms of pure TA, Compritol 888 ATO, the physical mixture of TA and Compritol 888 ATO, and TA loaded SLNs. Pure TA showed a sharp endothermic peak at 285.54°C corresponding to its melting point, indicating its characteristic crystalline nature. Compritol 888 ATO exhibits a sharp endothermic event, ascribing to the melting, around 74.52°C. These sharp melting endothermic peaks of bulk lipid indicate that the starting materials were crystalline. The peak for TA was completely absent in lyophilized TA loaded SLNs (), while it was clearly evident in the physical mixture of TA (285.54°C) and Compritol 888 ATO (74.52°C) as shown in . It has been reported that when the drug does not show its endothermic peak in the nanoparticulate formulations, it is said to be in the amorphous state (CitationLiu et al. 2005). Hence, it could be concluded that the drug was present in the amorphous phase and may have been homogeneously dispersed in the SLNs.
Figure 5. DSC thermograms. (a) Pure Compritol 888 (b) Pure TA (c) Physical mixture of TA and Compritol 888 ATO (d) TA loaded SLNs.
In vitro release study
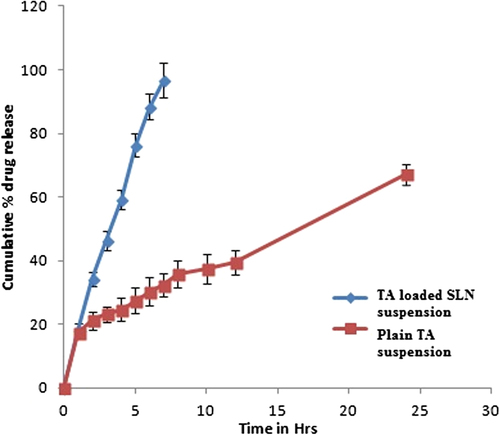
The cumulative percentage release of TA from TA suspension and TA loaded SLNs were investigated in vitro over a period of 24 h. Each sample was analysed in triplicate and release curves are shown in . It was indicated that plain TA suspension released almost 96.72 ± 5.46% of the drug at the end of 7 h, while in TA loaded SLNs 67.19 ± 3.23% drug release was observed after 24 h. TA loaded SLNs showed a biphasic drug release pattern with an initial burst release phase followed by a sustained drug release phase. TA loaded SLNs suspension depicted a biphasic release pattern characterized by relatively faster initial burst release followed by comparatively slower release with 67.19 ± 3.23% of TA released at 24 h. Burst release can be useful to improve the penetration of the drug, while sustained release supplies the drug for a prolonged period of time (CitationGupta et al. 2013). On the other hand, the drug release from plain TA suspension was faster with 96.72 ± 5.46% release of TA within 7 h. Regarding the drug release profiles, TA loaded SLNs suspension followed Higuchi release kinetics (r2 = 0.9909) and zero order release kinetics (r2 = 0.994), respectively. Similar results for novel and conventional formulation were obtained by CitationKhurana et al. (2013b).
Figure 6. Drug release profile from pure TA suspension and drug loaded SLNs suspension in phosphate buffer (7.4pH).
In vitro skin distribution study
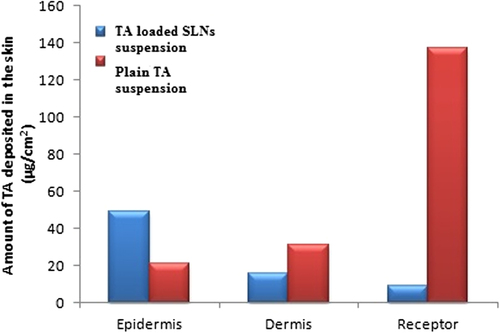
In vitro skin distribution study of TA loaded SLNs suspension and pure TA suspension were evaluated using goat skin. UV spectroscopy was used to quantify TA content in the epidermis, dermis and receptor. As shown in , plain TA suspension delivered maximum amount of TA in the receptor compartment (137.22 μg/ml) with minimum amount of TA in the epidermis (20.81/ml) and dermis (31.29 μg/ml) through 1 cm2 diffusion area indicating maximal distribution of drug in systemic circulation after 6 hrs. While TA loaded SLNs suspension delivered maximum amounts of TA (48.80 μg/ml) into the epidermal layer of skin with minimal amount of TA in the dermis (15.95 μg/ml) and in the receptor compartment (9.04 μg/ml) of the Franz cell, demonstrating that treatment with TA loaded SLNs would consequence into limited systemic escape of the steroidal drug. This systemic escape of the drug might eliminate adverse side effects associated with systemic exposure. In addition, the results of skin distribution study demonstrate that TA loaded SLNs suspension address the requirements of an efficient topical drug delivery system with minimal systemic escape and can be considered as an appropriate carrier for the delivery of TA in topical therapy of drugs having poor aqueous solubility, increased dosing frequency and dose dependant side effects. Based on the skin distribution data reported here, we hypothesize that TA delivery to the skin could be related to the increased solubility of drug in the lipid matrix (in case of SLNs) which in turn leads to an elevated concentration gradient at the skin surface rather than systemic circulation (CitationUner et al. 2014), thus, avoiding adverse side effects associated with the systemic circulation of drugs.
Figure 7. In vitro skin distribution study of pure TA suspension and TA loaded SLNs suspension.
Stability study
Stability studies were carried out according to CitationDas et al. (2011), to assess the stability and integrity of TA loaded SLNs. The TA loaded SLNs showed minor enhancement of the particle size and PI with slight reduction of ZP, EE, and L after 3 months storage at 4°C (). Percent change in PS and EE were 3.4% and 2.5%, respectively, on 3 months storage of SLNs formulations. The changes were insignificant which indicate good physical stability of the SLNs during their storage at 4°C for 3 months.
Table III. Stability study of selected formulation.
Conclusion
In this study, SLNs were successfully prepared by the emulsification–ultrasonication method. Compritol® 888 ATO demonstrated maximum TA solubilization capacity as compared with other lipids like Precirol® ATO5, glyceryl monostearate and stearic acid. Poorly water soluble drug TA was efficiently encapsulated into the solid lipid nanoparticles which were further confirmed by XRD and DSC study. Most of the process variables such as lipid concentration, surfactant concentration, drug concentration and sonication time showed significant effect on the formulation properties. The particle size remarkably increased with increasing lipid concentration and drug concentration while opposite tendency for the particle size was observed with increase in sonication time. The particle size of the selected batch was found to be 114.5 ± 1.32 nm. The drug release study exhibited prolonged release from the SLNs following Higuchi release kinetics (R2 = 0.9909) as compared with pure drug suspension (Zero order kinetics, R2 = 0.994). Stability study confirmed that SLNs were stable for 3 months at 4°C. Furthermore, in vitro skin distribution showed presence of significant amounts of TA into the epidermal layer of skin when treated with TA loaded SLNs suspension demonstrating the systemic escape of drug from TA loaded SLNs. Moreover, selective accumulation of TA in the epidermis might eliminate adverse side effects associated with systemic exposure. The encouraging results obtained in this study could propose this system for future in vivo studies, especially for delivery of other corticosteroids, useful in treatment of various dermal disorders.
Acknowledgement
The authors are thankful to the Director, University Institute of Pharmacy, Pt Ravi Shankar Shukla University Raipur, Chhattisgarh, for providing necessary infrastructural facilities, UGC-BSR for JRF (MP) and UGC-MRP-41-748-2012 for providing financial assistance relating to this work.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Charoenputtakhun P, Opanasopit P, Rojanarata T, Ngawhirunpat T. 2014. All-trans retinoic acid-loaded lipid nanoparticles as a transdermal drug delivery carrier. Pharm Dev Technol. 19:164–172.
- Chen H, Chang X, Du D, Liu W, Liu J, Weng T, et al. 2006. Podophyllotoxin-loaded solid lipid nanoparticles for epidermal targeting. J Control Release. 110:296–306.
- Das S, Ng WK, Kanaujia P, Kim S, Tan RBH. 2011. Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: effects of process variables. Colloids Surf B Biointerfaces. 88:483–489.
- Das S, Ng WK, Tan RB. 2012. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci 47:139–151.
- Ghadiri M, Fatemi S, Vatanara A, Doroud D, Najafabadi AR, Darabi M, Rahimi AA. 2012. Loading hydrophilic drug in solid lipid media as nanoparticles: statistical modeling of entrapment efficiency and particle size. Int J Pharm. 424:128–37.
- Gupta M, Tiwari S, Vyas SP. 2013. Influence of various lipid core on characteristics of SLNs designed for topical delivery of fluconazole against cutaneous candidiasis. Pharm Dev Technol. 18:550–559.
- Gupta V, Trivedi P. 2014. Ex vivo localization and permeation of cisplatin from novel topical formulations through excised pig, goat, and mice skin and in vitro characterization for effective management of skin-cited malignancies. Artif Cells Nanomed Biotechnol. Early Online: 1–10, doi:https://doi.org/10.3109/21691401.2014.893523.
- Hao J, Wang F, Wang X, Zhang D, Bi Y, Gao Y, et al. 2012. Development and optimization of baicalin-loaded solid lipid nanoparticles prepared by coacervation method using central composite design. Eur J Pharm Sci. 47:497–505.
- Agrawal Y, Petkar KC, Sawant KK. 2010. Development, evaluation and clinical studies of Acitretin loaded nanostructured lipid carriers for topical treatment of psoriasis. Int J Pharm. 401:93–102.
- Khurana S, Bedi PMS, Jain NK. 2013a. Preparation and evaluation of solid lipid nanoparticles based nanogel for dermal delivery of meloxicam. Chem Phy Lipids. 175:65–72.
- Khurana S, Jain NK, Bedi PMS. 2013b. Development and characterization of a novel controlled release drug delivery system based on nanostructured lipid carriers gel for meloxicam Life Sci. 93: 763–772.
- Lason E, Sikora E, Ogonowski J. 2013. Influence of process parameters on properties of Nanostructured Lipid Carriers (NLC) formulation. Acta Biochemica Polonica. 60:773–777.
- Liu J, Hu W, Chen H, Ni Q, Xu H, Yang X. 2007. Isotretinoin-loaded solid lipid nanoparticles with skin targeting for topical delivery. Int J Pharm. 328:191–195.
- Liu M, Dong J, Yang Y, Yang X, Xu H. 2005. Characterization and release of triptolide-loaded poly (d, l-lactic acid) nanoparticles. Eur Polym J. 41:375–382.
- Liu W, Hu M, Liu W, Xu CB, Xu H, Yang XL. 2008. Investigation of the carbopol gel of solid lipid nanoparticles for the transdermal iontophoretic delivery of triamcinolone acetonide acetate. Int J Pharm. 364:135–141.
- Lopes LB, Ferreira DA, Paula D, Garcia MJ, Thomazini JA, Fantini MA, Bentley MB. 2006. Reverse hexagonal phase nanodispersion of monoolein and oleic acid for topical delivery of peptides: in vitro and in vivo skin penetration of Cyclosporin A. Pharm Res. 23: 1332–1342.
- Mitri K, Shegokar R, Gohla S, Anselmi C, Müller RH. 2011. Lipid nanocarriers for dermal delivery of lutein: Preparation, characterization, stability and performance. Int J Pharm. 414:267–275.
- Mulla JS, Khazi IM. 2009. Influence of process variables on particle size of solid lipid nanoparticles. Ind J Novel Drug Deliv. 1:47–49.
- Patil GB, Patil ND, Deshmukh PK, Patil PO, Bari SB. 2014. Nanostructured lipid carriers as a potential vehicle for Carvedilol delivery: application of factorial design approach. Artif Cells Nanomed Biotechnol. Early online, doi:https://doi.org/10.3109/21691401.2014.909820.
- Pouton CW. 2006. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 29: 278–287.
- Pradhan M, Singh D, Singh MR. 2013. Novel colloidal carriers for psoriasis: current issues, mechanistic insight and novel delivery approaches. J Control Release. 170: 380–395.
- Prow TW, Grice JE, Lin LL, Faye R, Butler M, Becker W, et al. 2011. Nanoparticles and microparticles for skin drug delivery. Adv Drug Deliv Rev. 63:470–491.
- Rawat M, Saraf S, Saraf S. 2007. Influence of selected formulation variables on the preparation of enzyme-entrapped Eudragit S100 microspheres. AAPS Pharm Sci Tech. 4:289–297.
- Samein LH. 2014. Preparation and evaluation of nystatin loaded- solid-lipid nanoparticles for topical delivery. Int J Pharm Pharm Sci. 6:592–597.
- Schafer-Korting M, Mehnert W, Korting HC. 2007. Lipid nanoparticles for improved topical application of drugs for skin diseases. Adv Drug Deliv Rev. 59:427–443.
- Singh D, Dubey P, Pradhan M, Singh MR. 2013. Ceramic nanocarriers: versatile nanosystem for protein and peptide delivery. Expert Opin Drug Deliv. 10:241–259.
- Singh D, Nag M, Pradhan M, Singh MR. 2014. Vesicular system: versatile carrier for transdermal delivery of bioactives. Artif Cells Nanomed Biotechnol. Early Online, doi:https://doi.org/10.3109/21691401.2014.883401.
- Singh M, Singh D, Saraf S. 2011. Influence of selected formulation variables on the preparation of peptide loaded lipospheres. Trends Med Res 6:863–876.
- Singh MR, Pradhan K, Singh D. 2012. Lipid Matrix Systems with emphasis on lipid microspheres: Potent Carriers for Transcutaneous Delivery of Bioactives. Curr Drug Deliv. 9:243–254.
- Subedi RK, Kang KW, Choi H. 2009. Preparation and characterization of solid lipid nanoparticles loaded with doxorubicin. Eur J Pharm Sci. 37:508–513.
- Trotta M, Gallarate M, Pattarino F, Morel S. 2001. Emulsions containing partially water-miscible solvents for the preparation of drug nanosuspensions. J Control Release. 76:119–128.
- Uner M, Karaman EF, Aydoğmuş Z. 2014. Solid lipid nanoparticles and nanostructured lipid carriers of loratadine for topical application: physicochemical stability and drug penetration through rat skin. Tropical J Pharm Res. 13:653–660.
- Vitorino C, Carvalho FA, Almeida AJ, Sousa JJ, Pais AA. 2011. The size of solid lipid nanoparticles: an interpretation from experimental design. Colloids Surf B Biointerfaces. 84:117–130.