
Abstract
To develop an optimized supersaturable self-nanoemulsifying drug delivery system (S-SNEDDS) in order to improve the oral bioavailability of trans-resveratrol (t-RVT), together with surmounting poor aqueous solubility, enterohepatic recirculation and controlling drug precipitation, by employing a precipitation inhibitor (PPI) that is, hydroxypropyl methylcellulose (HPMC). The long-term stability of a previously reported formulation optimized long chain triglycerides (OPT LCT-SNEDDS), consisting of Lauroglycol FCC and Transcutol P, indicated rapid precipitation of trans-resveratrol. Following incorporation of the selected PPI, the precipitates were differentiated using the X-ray diffraction (XRD) technique. An in vitro supersaturation test was carried out for the S-SNEDDS formulation. The S-SNEDDS formulation was appraised for pharmacokinetic and in situ perfusion studies. In vitro dilution of the S-SNEDDS formulation resulted in the formation of a nanoemulsion, followed by a slow precipitation of t-RVT in the S-SNEDDS formulation vis-à-vis SNEDDS formulated with OPT-LCT, where it undergoes rapid precipitation, yielding a low t-RVT concentration. The pharmacokinetic study indicated that the AUC0-8h of the S-SNEDDS formulation increased by nearly 1.33-fold in the presence of HPMC vis-à-vis the OPT LCT-SNEDDS, at a drug dose of 20 mg/kg. The in situ perfusion parameters, viz., fraction absorbed and effective permeability, demonstrated significant improvement in the rate and extent of absorption from the S-SNEDDS formulation. This case demonstrates that the supersaturatable approach is effective in delivering and in improving the oral bioavailability of t-RVT.
Introduction
Trans-resveratrol (t-RVT) is a BCS class II drug with poor water-solubility and high permeability (log P of 3.1) (CitationAmri et al. 2012). Besides these, it undergoes rapid first-pass metabolism by CYP3A4 in the liver and suffers enterohepatic recirculation, eventually leading to a marked reduction in the drug's oral bioavailable fraction (almost zero) in humans and animals like rats, etc. (CitationAmri et al. 2012, CitationMarier et al. 2002, CitationSingh and Pai 2014a, Citation2014b).
To circumvent the aforementioned limitations, various formulations of t-RVT were developed, viz., using liposome, solid dispersions, β-cyclodextrin inclusion complex, microspheres and nanoparticles, but all with limited fruition (CitationHung et al. 2006, CitationWegiel et al. 2013, CitationTroche-Pesqueir et al. 2013, CitationNam et al. 2005, CitationKim et al. 2012). All these formulations were able to increase the solubility and/or dissolution performance only. We recently reported the use of PLGA nanoparticles for improvement in absorption and subsequent oral bioavailability of t-RVT (CitationSingh and Pai 2014a). CitationAmri et al. (2013) reported the self-emulsifying resveratrol drug delivery system, wherein they were able to achieve both the uptake by endothelial cells and improved protection against oxidative stress-mediated death. However, Amri et al. didn't address the problem of precipitation of the self-emulsifying drug delivery system. Thus, it can be clearly construed that none of these techniques were able to surmount the aforesaid problems.
Self-nanoemulsifying drug delivery systems (SNEDDS) are lipid-based nanocarriers that have recently exhibited an intriguing role in oral delivery of drugs, and have been reported to immensely augment the oral bioavailability by overcoming the above-mentioned problems (CitationElsheikh et al. 2012). Several potential advantages of SNEDDS include the capability of bypassing the hepatic portal route and promoting the lymphatic transport of lipophilic drugs, reducing metabolism by the cytochrome-P450 family of enzymes present in the gut enterocytes, and overcoming enterohepatic recirculation (CitationSingh and Pai 2014b; CitationBalakrishnan et al. 2009). By hypothesis, this was believed to be attained chiefly by lymphatic transport via Peyer's patches along the GI tract (CitationSingh and Pai 2014b).
The S-SNEDDS represent a newer technology containing a water-soluble polymeric precipitation inhibitor (PPI) intended to generate and maintain a meta-stable supersaturated state in vivo by preventing or minimizing the drug precipitation (CitationBandyopadhyay et al. 2014). Hydroxypropyl methylcellulose (HPMC) is the commonly used PPI in the S-SNEDDS. Of particular note is HPMC, which usually shows essentially more effectiveness in retarding drug precipitation and a consistently higher apparent concentration–time profile in comparison with other substances as PPI (CitationWei et al. 2012).
In continuation of the earlier research reported by us (CitationSingh and Pai 2014b), it was observed that with respect to the long-term stability, optimized long chain triglycerides (OPT LCT) SNEDDS underwent drastic changes in all the response parameters. Overall, the present research aims at developing S-SNEDDS of t-RVT based on our stability studies reported earlier, and the effect of excipients incorporated in the LCT-based SNEDDS formulation. In addition, the oral bioavailability of t-RVT from the S-SNEDDS formulation was also assessed, to determine whether S-SNEDDS may have any advantages in enhancing the oral bioavailability of t-RVT compared to the OPT LCT-SNEDDS.
Materials and methods
Materials
Trans-resveratrol was provided ex-gratis by M/s Sami Labs, Bangalore, Karnataka, India. Lauroglycol FCC and Transcutol P were received as gift samples from M/s Gattefosse, Mumbai, India. Methocel (HPMC E15LV) was purchased from M/s Sigma Aldrich, Mumbai, India. HPMC capsules (size 00) were obtained as generous gift samples from M/s ACG-Associated Capsules Pvt. Ltd, Mumbai, India. All other materials and chemicals procured for the studies were of analytical grade, and were used as such, as obtained.
Preparation of S-SNEDDS
The OPT LCT-SNEDDS formulation was prepared by a simple admixture method using Lauroglycol FCC (262 mg) and Transcutol P (225 mg), which we have reported earlier (CitationSingh and Pai 2014b). The PPI employed during the study was HPMC (E15LV). The PPI is incorporated in an amount equivalent to 5% of the total weight of the OPT LCT-SNEDDS formulation. The vials were vortexed vigorously for nearly 5 min to obtain a uniform suspension. The prepared formulation was stored at room temperature before use.
Apparent drug concentration–time profile
Solution samples (1 ml) of t-RVT from the S-SNEDDS formulation were taken without volume replacement at 5, 10, 15, 30, 45, 90, 120 and 180 min and filtered through a 0.45 μm filter. The filtered samples were suitably diluted with 100 ml of methanol (HPLC grade) followed by vortexing for about 2 min. The 20 μl filtrate was then assayed by high-performance liquid chromatography (HPLC) for determination of t-RVT, as we have reported earlier (CitationSingh et al. 2014). The apparent drug concentration–time profile and the duration of the supersaturated state were subsequently determined. A 100 ml of simulated gastric fluid (SGF) containing 0.01 M HCl and 0.15 M NaCl (pH 2.0) was added to a 250 ml round bottom flask equipped with a glass paddle. Experiments were conducted in triplicate and the medium was maintained at 37 ± 0.5°C, with the stirring speed held at 100 rpm.
Characterization of the S-SNEDDS formulation
Determination of globule size
An aliquot (1 ml) of the S-SNEDDS formulation, serially diluted 100-fold with purified water, was employed to assess the globule size using a particle size analyzer (Zetasizer S 90, M/s Malvern, Worcestershire, UK).
Characterization of precipitates
In vitro supersaturation test was carried out for the S-SNEDDS formulation and the OPT LCT-SNEDDS filled in HPMC capsules. Briefly, a 100 ml of SGF containing 0.01 M HCl and 0.15 M NaCl (pH 2.0) was added to a 200 ml round bottom flask equipped with a glass paddle, and the medium was maintained at 37 ± 0.5°C, with the stirring speed held at 100 rpm.
X-ray powder diffraction
In order to verify the crystallinity of the precipitates of both the S-SNEDDS and the OPT LCT-SNEDDS formulations, X-ray powder scattering measurements were carried out with an XRD-7000 diffractometer (Shimadzu Inc., Japan). The radiation source was generated by a Cu Kα radiation, with an applied voltage and current of 45 kV and 40 mA, respectively. The scanning speed was 10°/min from 3° to 50°, with a step size of 0.0185°.
In vivo pharmacokinetic study
The methodology for performing in vivo pharmacokinetic studies was similar to the method reported earlier by us (CitationSingh and Pai 2014b). All the animal investigations were performed as per the requisite protocol approved by the Institutional Animal Ethics Committee [Letter no AACP/IAEC/Jun-2012-02]. Briefly, one group of 6 male Wistar rats, weighing between 250 and 300 g, was taken S-SNEDDS formulation. All the animals received a dose equivalent to 20 mg of t-RVT per kg of body weight (CitationSingh and Pai 2014b).
Following oral drug administration, the rats kept in cages were allowed access to food and water, ad libitum. Serial aliquots of the blood samples (100 μl each) were withdrawn from the retro-orbital plexus, under mild ether anesthesia, at 0, 0.33, 0.5, 1, 3, 4, 6 and 8 h post-dosing, and placed in the heparinized microcentrifuge tubes (50 units heparin/ml of blood). Plasma was harvested by centrifugation at 15,000 × g for 15 min and stored at − 20°C until analyzed. Plasma samples were analyzed by HPLC. Non-compartmental pharmacokinetic parameters for extravascular input, that is, Cmax, Tmax, AUC, t1/2, Ke and MRT were computed by using the Kinetica 5.0.11 version software (Thermo Fisher Scientific Inc., Waltham, USA).
In situ single-pass intestinal perfusion studies
The single-pass intestinal perfusion (SPIP) study was carried out as per the method reported earlier by us (CitationSingh and Pai 2014b). Briefly, perfusion studies were performed in nine male Wistar rats (250–330 gm), previously made to abstain from solid food at least 24 h prior to the study. The SPIP studies for all the groups (i.e., pure drug, OPT LCT-SNEDDS and S-SNEDDS formulations) were carried out in triplicate, employing an in-house fabricated assembly. The intestinal segments were consequently perfused with pure drug, OPT LCT-SNEDDS and S-SNEDDS formulations, maintained at 37 ± 1°C, at a perfusion rate of 0.25 ml/min. Steady state was achieved within 30 min, after which aliquots of samples (1 ml each) were periodically withdrawn at regular intervals of 10 min each. The volume of sample for each time interval was 2 ml. The samples were stored at − 20°C until analysis. Samples were filtered and directly injected onto HPLC columns, and no sample preparation was required prior to analysis.
Stability studies
The S-SNEDDS formulation was subjected to stability studies, carried out at 25 ± 2°C/60% ± 5% RH, as per the ICH guidelines for the climatic zone IV. The formulation was kept in air-tight glass vials and assayed periodically, at the time points of 0, 1, 3 and 6 months, for percent drug release in 15 min and emulsification time.
Results
The purpose of formulating S-SNEDDS is to yield a supersaturated state, and as such, it is quite important to assess the drug concentration available in the supersaturated state, as well as the degree of supersaturation, as a function of time.
Characterization of S-SNEDDS formulation
Determination of globule size
During the globule size analysis of the S-SNEDDS formulation, it was observed that the polydispersity index (PDI) had increased to 0.468, thereby indicating a heterogeneous dispersion. In , three distinctive particle measurements (estimated from the intensity signal) were observed, with an approximate diameter of 357.5 nm (89.9%), 32.75 nm (5%) and 4457 nm (2.8%).
Figure 1. Globule size analysis of S-SNEDDS formulation.
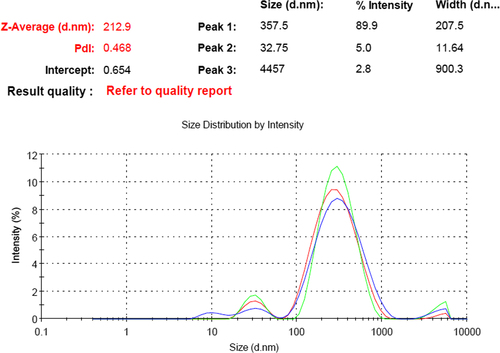
In vitro supersaturation test
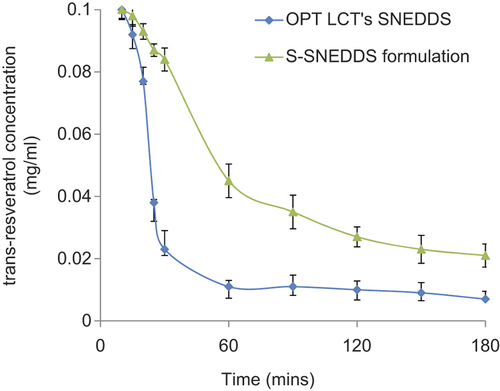
The OPT LCT-SNEDDS and S-SNEDDS formulations were evaluated by the supersaturation test (CitationBrouwers et al. 2009). Apparent t-RVT concentration–time profiles from the two formulations in the SGF (pH 2.0) are shown in . The apparent concentration of t-RVT in the test medium was 0.100 mg/ml. As shown in , the apparent concentration of t-RVT in the OPT LCT-SNEDDS was initially about 0.093 mg/ml at 10 min, which decreased rapidly to 0.011 mg/ml from 60 min onward, indicating the precipitation of t-RVT. The value of apparent concentration of t-RVT in the test medium of the OPT LCT-SNEDDS, over a 24 h course (i.e., equilibrium solubility), was estimated to be 0.009 mg/ml. By contrast, the S-SNEDDS formulation showed a consistently higher apparent concentration of t-RVT when compared with the OPT LCT-SNEDDS.
Figure 2. Apparent drug concentration–time profile of OPT LCT-SNEDDS and S-SNEDDS formulations.
X-ray powder diffraction
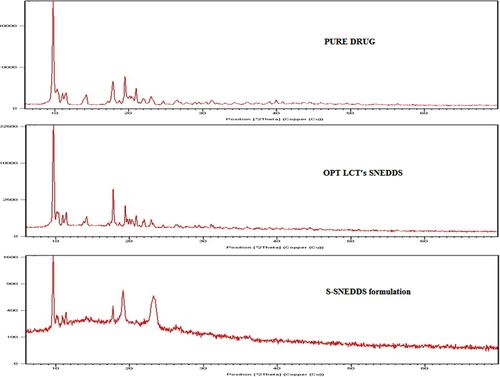
As shown in , t-RVT crystals exhibit characteristic sharp peaks as reported in previous works (CitationKim et al. 2012). The drug precipitates from OPT LCT-SNEDDS were highly crystalline, which can be confirmed from the pattern of the X-ray powder diffraction (XRPD) depicting sharp and intense peaks similar to the pure drug. By contrast, the precipitates collected from the S-SNEDDS formulation were amorphous, as the characteristic XRPD pattern of t-RVT was absent.
Figure 3. X-ray powder diffraction peaks of pure drug, OPT-LCT-SNEDDS and S-SNEDDS.
In vivo pharmacokinetic studies in rats
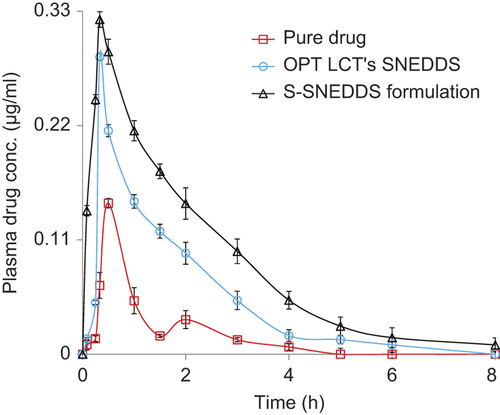
The mean plasma concentration–time profile () depicts significantly higher plasma levels in the S-SNEDDS formulation, with respect to pure drug and the OPT LCT-SNEDDS, at a t-RVT dose of 20 mg/kg (***p < 0.001). As both formulations showed rapid absorption with a short Tmax around 0.5 h, the S-SNEDDS formulation showed a significantly higher mean Cmax of 0.382 μg/ml vis-à-vis the pure drug and OPT LCT-SNEDDS. A nearly 1-fold augmentation in the magnitude of Ka was perceptible with the S-SNEDDS formulation, vis-à-vis the OPT LCT-SNEDDS.
Figure 4. Plasma drug level profiles of pure drug, OPT LCT-SNEDDS and S-SNEDDS formulations. Each point represents the mean of six replicates and each cross bar indicates 1 SEM. The data of two groups, that is, pure drug and OPT LCT-SNEDDS, have been reproduced with permission, from CitationSingh and Pai (2014b).
In situ intestinal perfusion studies
In the present studies, considerably higher magnitudes of absorptivity and permeability parameters were observed with the S-SNEDDS formulations vis-à-vis the OPT LCT-SNEDDS. The S-SNEDDS formulation showed a nearly 3.92 fold and 5.24 fold enhancement in effective and wall permeability respectively, vis-à-vis the pure drug. There was tremendous augmentation in the values of absorption number and fraction absorbed with the S-SNEDDS formulation, as compared to those of the pure drug. The increase in absorption number with the S-SNEDDS formulation was 3.84 fold, while the corresponding increase in the fraction absorbed was 3.03 fold. Overall, the results indicated that the S-SNEDDS formulation was remarkably superior to the pure drug in absorption and permeability.
Discussion
In an earlier report, we discussed the accelerated stability studies, wherein the OPT LCT-SNEDDS were found to be stable and robust. However, with respect to long-term stability, the OPT LCT-SNEDDS showed signs of turbidity after the 6th month of the study (at month-8). During the study, the values of all parameters (globule size, dissolution performance and emulsification time) had drastically changed at the 8th month, possibly due to drug precipitation. Gao and Morozowich have already reported the potential of S-SEDDS formulations in overcoming this hiccup, which subsequently established the role of S-SEDDS in improved bioavailability potential (CitationGao and Morozowich 2006).
It is reported that the hydrophobic HPMC is superior in sustaining a supersaturated state. Moreover, HPMC with a higher viscosity grade (i.e., a higher average MW), appears more effective in sustaining the supersaturated state (CitationWei et al. 2012). The roles of polymeric crystallization inhibitor have been reported in the last ten years. It is due to this difference in hydrophilicity that HPMC is more effective in the prevention of the nucleation process and the subsequent precipitation of t-RVT. These results are in consonance with literature findings, which report that significant enhancement in oral bioavailability of AMG 517 has been attained using the S-SNEDDS drug delivery system for improved oral absorption (CitationGao et al. 2009). Further, it was already reported that 5% of PPI is sufficient for retarding the drug precipitation, and the same is also evident from the results of the study (CitationGao et al. 2009). Hence, the findings clearly indicate that the presence of even a small amount of HPMC can significantly retard the t-RVT precipitation and sustain a higher apparent t-RVT concentration for approximately 90 min.
The PDI of the particle size was increased to 0.468, thereby indicating a heterogeneous dispersion. Owing to the heterogeneous distribution, globule size analysis was not feasible. Similar results were also observed with halofantrine-loaded S-SNEDDS (CitationThomas et al. 2012). Thus, dynamic light scattering (DLS) was practically not found to be a feasible method for size determination of the S-SNEDDS formulation. Further, it was reported that HPMC helps to form smaller sizes of emulsion, and prevents emulsion droplets from aggregation (CitationTadros 2009).
The droplet size characterization study demonstrated that the mean droplet size of the S-SNEDDS formulation was smaller than that of the OPT LCT-SNEDDS upon dilution with 0.1 M HCl, largely because of the presence of the HPMC.
Prototypes of the S-SNEDDS formulation and OPT LCT-SNEDDS (e.g., the same composition but without 5% HPMC) were evaluated by the supersaturation test. Upon mixing with the SGF, the OPT LCT-SNEDDS initially appeared slightly bluish in appearance. However, the solution developed cloudiness within 20 min and visible white solid precipitates of t-RVT were observed. Formation of t-RVT precipitates in the test medium suggested that the medium was in the supersaturated state. Similar observations were also reported for the molecules silybin and AMG 517 (CitationWei et al. 2012, CitationGao et al. 2009). The results indicate that the presence of a small amount of HPMC (5%, w/w) can retard t-RVT precipitation and sustain a higher apparent concentration over approximately 60 min.
These results imply that HPMC effectively interferes with the nucleation process of t-RVT, thus sustaining a supersaturated state of t-RVT. The general applicability of cellulosic polymers in inhibiting crystallization of many pharmaceutical substances has already been widely reported (CitationThomas et al. 2012). Conclusively, the results from the above studies suggest that t-RVT was in an amorphous or molecular dispersion state when administered as an S-SNEDDS formulation, thereby controlling its precipitation. These results, which are in good agreement with the in vitro results described above, imply a higher free drug concentration in the GI lumens, resulting from the supersaturated state, as desired. Both the in vitro test of supersaturation and the in vivo pharmacokinetic performance clearly indicate that the S-SNEDDS formulation is remarkably effective in suppressing macroscopic precipitation of t-RVT, thus improving the rate and extent of absorption of t-RVT.
The low values of AUC and Cmax after administration of an adequate dose of pure drug (20 mg/kg) also point toward incomplete absorption of the drug in rats. The values of Tmax, that is, 0.5 h observed for pure drug in rats, are quite in accordance with literature (CitationSingh and Pai 2014a, Citation2014b). This low absorption rate potential can be attributed to slower and incomplete dissolution of this BCS class II drug in the GI tract. Thus, besides poor aqueous solubility of t-RVT, the decreased bioavailability in rats may be due to a multitude of other reasons. The potential causes for decreased absorption include thicker GI barriers, higher hepatic first-pass metabolism, enterohepatic circulation, lower residence time in the GI tract and lower absorptive surface area.
The higher Cmax levels, coupled with the higher values of AUC observed in the S-SNEDDS formulation, indisputably vouch for distinct improvement in rate and extent of drug bioavailability. This can possibly be attributed to the lymphatic transport of t-RVT by such systems. This augmentation in bioavailability would eventually result in an escalation in the intensity of therapeutic effect of t-RVT. Exclusively higher values of absorption rate constant (Ka) and lower values of Tmax for the S-SNEDDS formulation vis-à-vis the pure drug indicate significantly quicker absorption of t-RVT from the S-SNEDDS and reduced precipitation of the drug.
The second peak at 2 h indicates enterohepatic recirculation of the drug, which has been well documented, and enterohepatic recirculation is susceptible to circadian variation (as is evident from , where two peaks of Cmax and Tmax were observed) (CitationAlmeida et al. 2009). A major portion of the bile acids secreted is reabsorbed from the intestines and returned via the portal circulation to the liver, thus completing the enterohepatic cycle. The parent compound and its conjugated metabolite undergo enterohepatic recirculation, resulting in multiple peaks in the plasma concentration–time profile (CitationSingh and Pai 2014c).
Marked enhancement in the effective permeability, a direct measure of absorption of the S-SNEDDS across the intestine, clearly demonstrates that the S-SNEDDS formulation was able to augment drug absorption in rats. In addition, bypassing the portal circulation also avoids further metabolism in liver and enterohepatic circulation (CitationSingh and Pai 2014b).
The in situ SPIP technique is useful for studying mechanisms of absorption and regional disposition of drugs. In this regard, SPIP studies are documented for their utility in augmenting the absorption and permeation potential of a drug when administered through self-nanoemulsifying systems. Thus, the results clearly indicate the ability of the SNEDDS formulation in augmenting the rate and extent of absorption, thereby showing promise in augmenting the oral bioavailability of t-RVT. The wall permeability provides an idea about the uptake mechanisms across the intestinal epithelium (CitationJohnson and Amidon 1998). SNEDDS, being primarily a carrier-based system, is actively transported across the GI via carrier-mediated transport (CitationSingh and Pai 2014b). Thus, the S-SNEDDS have been able to deliver the drug via the transcellular route, thereby achieving increased degree of lymphatic targeting (CitationPorter and Charman 2001). Overall, the S-SNEDDS were able to increase the absorption and permeation, demonstrating their potential in augmenting the oral bioavailability of t-RVT.
Parameters during the stability studies unambiguously show that the globule size, dissolution performance and emulsification time of the S-SNEDDS formulation were negligibly altered during 6 months of storage under stable conditions, ratifying the high robustness of the formulation.
Conclusion
The results indicate that in order to augment the oral bioavailability of t-RVT, an S-SNEDDS formulation containing a small amount of cellulosic polymer (i.e., HPMC) as a precipitation inhibitor was designed. Considerable augmentation in the rate and extent of oral drug absorption ratified the better performance of the S-SNEDDS formulation in enhancing the bioavailability of t-RVT. Further, SPIP reveals the significant utility of the S-SNEDDS formulation in augmenting the systemic exposure and subsequent bioavailability potential of t-RVT. Conclusively, the work demonstrates the critical importance of the supersaturated state in increasing the drug solubility in intestinal fluids, facilitating enhanced oral absorption and permeation across the GI tract. Thus, S-SNEDDS is a promising way to deliver t-RVT by the oral route.
Acknowledgements
The authors would like to thank Prof. B.G Shivananda for his advice and support in carrying out this research. The authors gratefully acknowledge the financial support and the research fellowship grant (45/38/2011/Nan-BMS) from ICMR (Indian Council of Medical Research, Government of India, New Delhi). The authors are also grateful to Sami Labs, Bangalore, India, for providing the gift sample of trans-resveratrol.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Almeida L, Vaz-da-Silva M, Falcao A, Soares E, Costa R, Loureiro AI, et al. 2009. Pharmacokinetic and safety profile of trans-resveratrol in a rising multiple-dose study in healthy volunteers. Mol Nutr Food Res. 53:S7–15.
- Amri A, Chaumeil JC, Sfar S, Charrueau C. 2012. Administration of resveratrol: What formulation solutions to bioavailability limitations. J Control Release. 158:182–193.
- Amri A, Le Clanche S, Thérond P, Bonnefont-Rousselot D. 2013. Resveratrol self-emulsifying system increases the uptake by endothelial cells and improves protection against oxidative stress-mediated death. Eur J Pharm Biopharm doi:https://doi.org/10.1016/j.ejpb.2013.10.015.
- Balakrishnan P, Lee BJ, Oh DH. 2009. Enhanced oral bioavailability of dexibuprofen by a novel solid Self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm. 72:539–545.
- Bandyopadhyay S, Katare OP, Singh B. 2014. Development of optimized supersaturable self-nanoemulsifying systems of ezetimibe: effect of polymers and efflux transporters. Expert Opin Drug Deliv. 11:479–492.
- Brouwers J, Brewster ME, Augustijns P. 2009. Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci. 98:2549–2572.
- Elsheikh MA, Elnaggar YSR, Gohar EY, Abdallah OY. 2012. Nanoemulsion liquid preconcentrates for raloxifene hydrochloride: optimization and in vivo appraisal. Int J Nanomedicine. 7:3787–3802.
- Gao P, Morozowich W. 2006. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin Drug Deliv. 3:97–110.
- Gao P, Akrami A, Alvarez F, Hu J, Li L, Ma C, Surapaneni S. 2009. Characterization and optimization of AMG 517 supersaturatable selfemulsifying drug delivery system (S-SEDDS) for improved oral absorption. J Pharm Sci. 98:516–528.
- Hung CF, Chen JK, Liao MH, Lo HM, Fang JY. 2006. Development and evaluation of emulsion-liposome blends for resveratrol delivery. J Nanosci Nanotechnol. 6:2950–2958.
- Johnson DA, Amidon GL. 1998. Determination of intrinsic membrane transport parameters from perfused intestine experiments: a boundary layer approach to estimating the aqueous and unbiased membrane permeabilities. J Theor Biol. 131:93–106.
- Kim S, Na WK, Dong Y, Das S, Tan RBH. 2012. Preparation and physicochemical characterization of trans-resveratrol nanoparticles by temperature-controlled antisolvent precipitation. J Food Eng. 10:37–42.
- Marier JF, Vachon P, Gritsas A. 2002. Metabolism and disposition of resveratrol in rats: extent of absorption, glucuronidation, and enterohepatic recirculation evidenced by a linked-rat model. J Pharmacol Exp Ther. 302:369–373.
- Nam JB, Ryu JH, Kim JW, Chang IS, Suh KD. 2005. Stabilization of resveratrol immobilized in monodisperse cyano-functionalized porous polymeric microspheres. Polymer. 46:8956–8963.
- Porter CJ, Charman WN. 2001. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev. 50:61–80.
- Singh G, Pai RS. 2014a. Optimized PLGA nanoparticle platform for orally dosed trans-resveratrol 380 with enhanced bioavailability potential. Expert Opin Drug Deliv. 11:647–659.
- Singh G, Pai RS. 2014b. Trans-resveratrol self nano-emulsifying drug delivery systems 336 (SNEDDS) with enhanced bioavailability potential: Optimization, pharmacokinetics and in situ single pass intestinal perfusion (SPIP) studies. Drug Deliv. doi:https://doi.org/10.3109/10717544.2014.885616.
- Singh G, Pai RS. 2014c. Recent advances of resveratrol in nanostructured based delivery systems and in the management of HIV/AIDS. J Control Release. 194:178–188.
- Singh G, Pai RS, Pandit V. 2014. In vivo pharmacokinetic applicability of a simple and validated HPLC method for orally administered trans-resveratrol loaded polymeric nanoparticles to rats. J Pharm Investig. 44:69–78.
- Tadros T. (2009). Polymeric surfactants in disperse systems. Adv Colloid Interface Sci. 147–148:281–299.
- Thomas N, Holm R, Mullertz A, Rades T. 2012. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 160: 25–32.
- Troche-Pesqueir E, Perez-Juste I, Navarro-Vazquez A, Cid MM. 2013. A β-cyclodextrin–resveratrol inclusion complex and the role of geometrical and electronic effects on its electronic induced circular dichroism. RSC Adv. 3:10242–10250.
- Wei Y, Ye X, Shang X, Peng X, Bao Q, Liu M, et al. 2012. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf A Physicochem Eng Asp. 396:22–28.
- Wegiel LA, Mauer LJ, Edgar KJ, Taylor LS. 2013. Crystallization of amorphous solid dispersions of resveratrol during preparation and storage-Impact of different polymers. J Pharm Sci. 102:171–184.