
Abstract
The purpose of this research was to develop a suitable and alternate adjuvant for the tetanus toxoid (TT) vaccine that induces long term immunity after a single-dose immunization. In our study, the preformulation studies were carried out by using different ratios (7/3, 8/2, and 9/1) of chitosan–hydroxypropyl methylcellulose (HPMC)-blended empty microspheres. Moreover, TT was stabilized with heparin (at heparin concentrations of 1%, 2%, 3%, and 4% w/v) and encapsulated in ideal chitosan – HPMC (CHBMS) microspheres, by the water-in-oil-in-water (W/O/W) multiple emulsion method. The vaccine entrapment and the in vitro release efficiency of the CHBMS was evaluated for a period of 90 days. The release of antigens from the microspheres was determined by ELISA. Antigen integrity was investigated by SDS-PAGE. From the optimization studies, it was found that a chitosan/HPMC ratio of 8/2 produced a good yield, with microspheres that were spherical, regular and uniformly-sized. In the CHBMS, a heparin concentration of 3% w/v resulted in well-sustained antigen delivery for a period of 90 days. It was found that the characteristics of initial release could be observed in 2 days, followed by a constant release, and an almost 100% complete release in 90 days. From the in vitro release characteristics, the ideal batch of CHBMS (3% w/v heparin) was evaluated for in vivo studies by the antibody induction method. The antibody levels were measured for different combinations for the period of 9 months, and finally, with a second booster dose after 1 year. In conclusion, it was observed that CHBMS (combination-1) resulted in the antibody level of 4.5 IU/mL of guinea pig serum, and the level was 3.5 IU/mL for the Central Research Institute's alum-adsorbed tetanus toxoid (CRITT) (combination 2), after 1 year, with a second booster dose. This novel approach of using CHBMS may have potential advantages for single-step immunization with vaccines.
Introduction
The main aim of the World Health Organization (WHO) is to develop efficient and inexpensive vaccines against various infectious diseases. Tetanus is a rare disease in most industrialized countries, but it is still a significant problem concerning public health in many parts of the developing world. It kills over 4 lakh people annually (CitationOladiran et al. 2002). Clostridium tetani is a spore-forming, Gram-positive bacillus (CitationVandelaer et al. 2003). The requirement for multiple injections of the currently licensed tetanus vaccine dictates that there is often comparatively poor coverage in countries where economic or logistical factors preclude this. Therefore, the WHO has emphasized that the development of improved immunization tactics to control diseases is a priority (CitationAlpar et al. 1994). Traditionally, adjuvants have been used in vaccine formulations to augment the immune response, usually after a booster dose. Aluminum phosphate and aluminum hydroxide are the approved adjuvants for human vaccination and are widely used for vaccines at present (CitationGupta and Siber 1995). In recent years, there have been various attempts to demonstrate new and improved techniques to induce a higher level of immunogenicity following parenteral and/or oral administration (CitationKohn et al. 1986).
Currently, vaccine development is experiencing another wave of tremendous progress, with the aid of modern immunology and biotechnology. More recently, the children's vaccine initiative was established to promote the development of new vaccines that could be administered early in children's lives, would require fewer doses, would not require refrigerated storage and would have simple immunization schedules through easier routes of administration (CitationO’Hagan et al. 1991). Therefore, the development of a more efficient and safe vaccine delivery system requiring a single administration to obtain high and long lasting response, is of importance. Various vaccine targets have been identified, besides prophylactic diseases; for example, vaccine development has also been carried out for cancer and acquired immune deficiency syndrome (AIDS) (CitationZhao and Leong 1996). One approach has been to use biodegradable polymer technology, to simplify a sustained-release antigen following parenteral administration, which, it is envisaged, will minimize the injections given in the normal regimen (CitationAlpar et al. 1994). Moreover, several studies have indicated the potential of biodegradable microparticles to enhance the immunogenicity of poorly immunogenic molecules (CitationSingh et al. 1997) and specific vaccines (CitationEldridge et al. 1989).
Controlled drug delivery technology using biodegradable polymers as carriers represents one of the most rapidly advancing areas of science. Controlled delivery systems consisting of biodegradable microspheres can potentially deliver either the antigens or adjuvants to the desired location, at predetermined rates and durations, to generate an optimal immune response. The carrier may also protect the vaccine from degradation until it is released. Other potential advantages of the controlled delivery approach include reduced systemic side effects, and the possibility of coencapsulating multiple antigenic epitopes or both antigen and adjuvant in a single carrier. Although the currently available vaccines represent an outstanding success story in modern medicine and have had a dramatic effect on morbidity and mortality worldwide, it is clear that improvements are required in the current technologies for vaccine delivery to control diseases, over the conventional methods of approach (CitationDerek et al. 2004).
Chitosan is derived by the deacetylation of chitin, which is a polymer of D-glucosamine and N-acetyl D-glucosamine. Chitosan is well known for its hydrophilic, biocompatible, biodegradable and non-toxic properties (CitationIllum 1998, CitationFelt et al. 1998, CitationJoane et al. 1999). Chitosan suspensions or microparticles and nanoparticles have been reported to have immune stimulating activity, such as increasing the accumulation and activation of macrophages and polymorphonuclear cells, promoting resistance to infections by microorganisms, and inducing cytokines (Citationvan der Lubben et al. 2001). In particular, regarding vaccines, the conversion of multiple-dose vaccines into single-dose vaccines represents an important advancement towards the betterment of human health care (CitationCleland et al. 1996).
HPMC is also a carbohydrate polymer, soluble in water. It forms aqueous solutions when heated above a particular temperature and exhibits a unique property of forming reversible physical gels due to hydrophobic interactions (CitationBilati et al. 2005). HPMC finds applications as a binder or thickener in pharmaceutical, food and ceramic processing industries, and can undergo thermo-reversible gelation in an aqueous solution upon heating (CitationHirrien et al. 1996, Citation1998, CitationIbbett et al. 1992).
Here, we studied the preformulation of empty microspheres with different ratios of chitosan/HPMC (7/3, 8/2 and 9/1), and the effect of using heparin (concentrations of 1%, 2%, 3%, and 4% w/v) as stabilizer, on in vitro release characteristics. Based on the preformulation and the in vitro release patterns, the ideal batch of microspheres was used to measure the antibody levels in guinea pigs, by comparing with CRITT.
Materials and methods
Chitosan (viscosity grade 150 cps, 80% deacetylation) was purchased from the Central Institute of Fisheries Technology (Cochin, India). Tween 80 was purchased from Fluka (Buchs, Switzerland). Tetanus toxoid having a concentration 1250 Lf/ml and the standard tetanus antitoxin were received as gifts from the Central Research Institute (Kasauli, H.P, India). HPMC, heparin, bovine serum albumin (BSA), and sodium tripolyphosphate (TPP) were purchased from Sigma Aldrich (St. Louis, MO, USA). All other chemicals and reagents used were of analytical grade.
Preparation of chitosan–HPMC-blended empty microspheres
The W/O/W multiple emulsion technique was used to prepare the CHBMS by following the procedure, with slight modifications (CitationJameela et al. 1994). Three batches of microspheres were prepared by altering the ratio of chitosan/HPMC (7/3, 8/2 and 9/1). The dispersion phase was prepared by mixing 50 ml of linseed oil and 10% of Tween 80 at 2000 rpm for 10 min. To this, 3 ml of various ratios of chitosan/HPMC were mixed separately to form a water-in-oil emulsion (W/O). At the end of the second hour, 2 mL of TPP (5% w/v) was separately introduced dropwise and the stirring was continued for another 2 h, followed by the addition of another 2 mL of TPP. Finally, the suspension of microspheres was centrifuged at 15,000 rpm to remove the extra oil, and the pellets were washed with toluene and acetone. The dried microspheres were lyophilized and stored in a sealed glass vial in a vacuum desiccator.
Preparation of TT-encapsulated CHBMS
The CHBMS were prepared by the same method as for chitosan–HPMC- blended empty microspheres, but the TT (2 mL, 1250 Lf units/mL) was introduced dropwise after formation of the W/O emulsion. Four batches of CHBMS were prepared separately by varying the concentration of the stabilizer, heparin (1%, 2%, 3%, and 4% w/v).
Morphological observations
The morphological examination of the empty microspheres and the CHBMS were performed using a scanning electron microscope (SEM; JSM T-220 A, Jeol, Japan). The microspheres were mounted on to metal stubs using double-sided carbon adhesive tape. After being vacuum-coated with gold palladium to a thickness of 200–300A°, the microspheres were examined with the SEM at 4 kV.
Estimation of TT content in CHBMS
Ten mg of CHBMS was powdered in an agate mortar and the TT was extracted with 6 mL of phosphate-buffered saline (PBS) (pH 7.4) in screw-capped vials, by rotating the tubes head to tail in an hematology mixer for 7 days (CitationBarman et al. 2000). The total antigen content in the extract was determined by ELISA. Complete extraction was obtained by further treating the microspheres with 5% v/v hydrochloric acid in ethanol again, for a further 2 days. Blank microspheres with heparin were used as control.
Estimation of antigen integrity
The extracted TT antigen solution was run on SDS-PAGE gel (Biotech R & D Laboratories, Yercaud), using standard protocols. A gel was prepared with stacking and separating gels of 4% and 8% polyacrylamide, respectively. Equal concentrations of samples were loaded carefully into the gel wells with the help of a micropipette, and it was run at a constant voltage (150 V) until the dye band reached the bottom of the gel. The gel was stained with a 0.1% Coomassie Blue fixative solution, and then destained using an aqueous solution containing 40% (v/v) methanol and 10% (v/v) acetic acid.
In vitro release study
In vitro release studies of the prepared microspheres with various concentrations of heparin (1%, 2%, 3% and 4% w/v) were carried out separately in a conical flask, by taking 50 mg of microspheres and 50 ml of PBST (pH 7.4), which were incubated at 37°C on a constant shaking mixer (CitationBarman et al. 2000). The content of the vial was withdrawn and centrifuged at 5000 rpm for 5 min at predetermined time intervals (days 0, 2, 4, 8, 12, 16, 21, 28, 35, 42, 49, 56, 63, 70, 77, 84 and 90). The TT concentrations in the supernatants were determined by the ELISA method. Placebo microspheres without antigen were used as control.
ELISA for TT
The antigenicity of TT was measured by ELISA, as previously described, with slight modifications (CitationJohansen et al. 1998). Briefly, flat-bottom 96-well immune microtiter plates (NUNC) were filled with 100 μl of 2 IU/mL of horse anti- tetanus IgG in 0.05 M carbonate buffer of pH 9.6, overnight. The plates were washed three times with 300 μl of PBS containing 0.05% Tween 20. After this, a two-fold dilution series of sample and reference TT samples were prepared using PBS with 0.5% BSA. The plates were held at room temperature for 24 h, followed by the addition of peroxidase-labelled sheep anti-TT serum (in PBS with 0.5% BSA) at room temperature, for 2 hours. Finally, 100 μl of 0.2 mg/mL substrate solution of 2, 2’-azino-bis (3-ethyl-benzthiazoline-6-sulfonate ABTS® Peroxidase) in 100 mM NaH2PO4 solution, was added to the plates. The reaction was stopped by the addition of 5 N H2SO4, at 50 μl/well. The plates were read on an ELISA reader (VersaMax tunable microplate reader, Sunnyvale, California, USA) after 30 min, at a wavelength of 405 nm.
In vivo immunogenicity study
The ideal batch of CHBMS (3%, w/v of heparin) encapsulated with TT was tested for immunizing potency, and the antibody response to tetanus was measured in international units (IU) per mL of serum. The grouping and vaccine combinations used for the immunization of the guinea pigs, either with the booster dose or without the booster dose, have been categorized in . The animal studies were carried out based on the recommendations by the Institutional Animal Ethics Committee of the Central Research Institute (CRI).
Table I. Grouping and combinations used for in vivo immunogenicity study (n = 9).
Six groups of nine guinea pigs (250–350 g) for each group were immunized with the combination of vaccines. The booster dose, wherever indicated, was given not later than 4 weeks from the date of primary immunization. Bleeding was done at about 2 weeks after the booster dose. The sera of the immunized guinea pigs were collected and tested in mice and guinea pigs respectively, for the measurement of tetanus antibodies. The potency test was conducted according to the standards of the National Control Laboratory (CRI, Kasauli) and the CitationIndian Pharmacopoeia (1985). Bleeding of the immunized guinea pigs was done by cardiac puncture using a 20 gauge needle. The blood samples were collected in sterile glass tubes and kept in a slanting position, so as to allow the serum to ooze out of the clotted bottle. Then, these tubes were centrifuged at 2000 rpm for 45 min. The supernatant immune serum from each tube was transferred to another sterile 5 mL vial using sterile Pasteur pipettes, and numbered. The sera samples were inactivated at 55°C in a water bath for 20 min, before they were tested for tetanus antibodies by the antibody induction method.
Results
Morphological characterization of empty and CHBMS microspheres
Three batches of chitosan–HPMC empty microspheres (7/3, 8/2 and 9/1) were prepared, and the morphological and size characteristics were evaluated. The microspheres with chitosan/HPMC ratios of 7/3 and 9/1 were observed to have an irregular shape, with some clumping and crystals (). In the ratio of 8/2, spherical, regular and uniformly-sized microspheres of 10 μm size were observed (). The size of all the prepared microspheres was in the range between 1 and 40 μm. Microspheres prepared less than 10 μm in size are reportedly taken up by antigen- presenting cells and thus considered to be the most suitable for optimum adjuvant effect (CitationEldridge et al. 1991, CitationO’Hagan et al. 1993).
Figure 1. Effect of morphology on SEM image of chitosan-HPMC blended empty microspheres. (A) 7/3, (B) 9/1, (C) 8/2.

In CHBMS, the stabilized TT (heparin, 1%, 2%, 3%, and 4% w/v) was encapsulated with microspheres prepared with an 8/2 ratio of chitosan/HPMC. The size of the particles was equal to or less than 10 μm, and they were observed to have more uniform distribution, no roughness, no porosity, no clumping, and high sphericity (). The encapsulation efficiency of both CHBMS with heparin and without heparin was measured by ELISA. The TT payload of CHBMS with heparin varied from 70% to 80%, whereas in the CHBMS without heparin, a payload of only 30% – 40% was observed.
Figure 2. SEM image of TT-encapsulated chitosan–HPMC microspheres.

Antigen integrity
The antigen integrity of TT microspheres with or without the stabilizer, heparin, was investigated by SDS-PAGE, followed by Coomassie Brilliant Blue staining. The antigen integrity of TT encapsulated in the chitosan – HPMC microspheres with heparin as the stabilizer, remained undamaged, as shown in . However, the TT encapsulated without the stabilizer was altered, which may be due to protein aggregation during emulsification. These observations clearly indicated that the protein stabilizer; heparin, plays a major role in the encapsulation of TT, without inactivation or aggregation in to the chitosan-HPMC microspheres (CitationCleland and Jones 1996).
Figure 3. SDS-PAGE analysis of TT. Lane 1: standard TT, Lane 2: TT extracted from chitosan–HPMC microspheres without heparin, Lane 3: TT extracted from chitosan–HPMC microspheres.

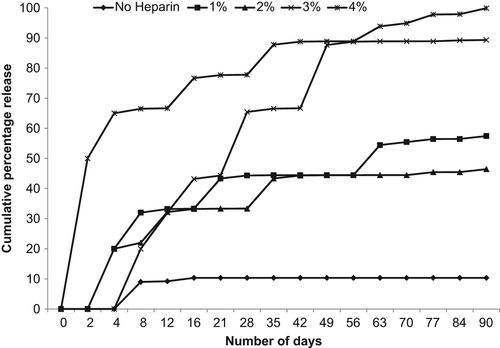
In vitro release study of CHBMS
The in vitro release of CHBMS stabilized with heparin at different concentrations (1%, 2%, 3%, and 4% w/v) was compared with microspheres prepared without the heparin stabilizer. In CHBMS prepared without heparin, the maximum release observed was only 10%. This could be the inactivation or aggregation of antigen in the first emulsion step. Therefore, the best approach to avoid aggregation is the addition of a suitable stabilizer, which will protect the TT from organic solvents, by preferential hydration of their surface, thus preventing the protein-interface contact (CitationCleland and Jones 1996). With a heparin concentration of 4% w/v, the initial burst was increased, and almost 70% was released within 15 days of incubation. However, in the microspheres prepared with 3% w/v of heparin, 7–10% was released in the first 2–5 days, and sustained release was observed thereafter for 90 days (). All the remaining batches showed irregular or lower initial release. From the in vitro release studies, we concluded that the protein stabilizer, heparin, prevents the denaturation of TT in the first emulsion step, increasing the payload, which was reflected in the increased and sustained cumulative percentage release.
Figure 4. Cumulative percentage release profiles of TT encapsulated in to chitosan–HPMC microspheres with or without heparin.
In vivo immunogenicity study
The level of specific antibody against tetanus, present in the immune guinea pig serum collected from guinea pigs immunized with CHBMS (3% w/v of heparin) and adsorbed on to CRITT, is given in .
Table II. The antibody levels of immune guinea pig sera after first bleeding at 6 months.
The administration of CHBMS to nine guinea pigs as primary immunization with one booster dose, gave raise to immune sera with a maximum antibody level of 3.5 IU/mL of guinea pig serum. When no booster was given, the level of antibody was found to be 1.5 IU/mL of guinea pig serum. The sera samples collected from guinea pigs primarily immunized with alum-adsorbed TT (CRITT) and given a booster dose of CHBMS, contained a maximum antibody level of 2 IU/mL of guinea pig serum. Similarly, the primary immunization done with CHBMS and CRITT as booster dose also gave rise to an antibody level of 2 IU/mL of guinea pig serum. When the control CRITT was given for primary and booster inoculations, the antibody level in the immune sera was found to be 2.5 IU/mL of guinea pig serum. The antibody level was found to be only 1 IU/mL of guinea pig serum when no CRITT booster was given.
As per the I.P. requirement, all the combinations of primary immunization and booster dose passed the potency test as the minimum requirement with at least 2/3rd of the guinea pig sera containing a minimum antibody level of 0.05 IU/mL of guinea pig serum and at least 1/3rd of the guinea pig sera containing an antibody level of 0.5 IU/mL of guinea pig serum.
After 9 months, the same guinea pigs were again bled and the sera were estimated for specific antibody level. In case of combination-1, the guinea pig sera still continued to give a maximum of 2.5 IU/mL of guinea pig serum. However, in case of combination-2, the maximum potency unitage was found to be 2 IU/mL of guinea pig serum. These findings show that after a period of 9 months, there is a gradual decline in the tetanus antibody levels. The results were shown in .
Table III. Antibody levels of immune guinea pig sera 9 months after primary immunization.
The second booster dose was given after 9 months, and the antibody levels were measured after 1 year. In the case of CHBMS (combination-1), the antibody level rose up to 4.5 IU/mL of guinea pig serum. In the case of the control TT-adsorbed (CRITT) combination-2, after the 2nd booster dose, the potency unitage was found to be 3.5 IU/ml of guinea pig serum, which is not even comparable to the result obtained with combination-1. The results are shown in . The in vivo results show that the antibody levels of combination-1 were higher at months 6, 9 and 12 following primary and booster immunizations.
Table IV. Antibody levels of immune guinea pig sera 1 year after primary immunization with second booster dose.
Discussion
An adjuvant is an agent that stimulates or activates the immune system, increasing the response to a vaccine, while not having any specific antigenic property. Adjuvants are immune stimulating materials that perform the function of a “depot” for the antigen, target the antigen to immune cells (APCs), and enhance phagocytosis and modulation of immune responses induced by the antigen alone (CitationCox and Coulter 1997, CitationTrujillo Vargas et al. 2005, CitationLutsiak et al. 2006, CitationPetrovsky and Aguilar 2004). Aluminum containing compounds such as aluminum hydroxide or aluminum phosphate are the adjuvants most widely used in human vaccines. Compared with the other adjuvants, alum is relatively weak (CitationKreuter and Speiser 1976) and may not be able to sufficiently enhance the immune response towards a poorly immunogenic antigen. Alum can induce the formation of a granuloma at the site of injection (CitationWarren et al. 1986) and stimulate the production of IgE antibodies, which are responsible for mediating immediate hypersensitive reactions.
Therefore, there is an urgent need to develop new adjuvants with minimum or no side effects. In our present study, we attempted to develop a single-shot vaccine delivery using CHBMS. In the preformulation studies, we prepared three batches of microspheres, with chitosan/HPMC ratios of 7/3, 8/2 and 9/1, by the emulsion cross-linking technique. The results showed that the ratio of 8/2 produced regular, spherical, uniformly sized particles (10 μm) and the ratios of 7/2 and 9/1 resulted in highly clumped, irregular microspheres (> 10 μm). This may be due to aggregation or swelling of polymers and the fact that microspheres which are more than 10 μm in size are not taken up by the antigen-presenting cells to provide a good adjuvant effect. Therefore, we selected the 8/2 ratio for preparation of the TT-encapsulated CHBMS.
Our previous studies on the in vitro release characteristics of single or composite polymeric microspheres stabilized with sucrose or lactose reported sustained delivery of the antigen for 70 days (CitationArthanari et al. 2011, Citation2014). Here, we confirmed that the CHBMS stabilized with heparin extends the release of TT for 90 days. To prove this, we developed four batches of microspheres with different concentrations of heparin. A heparin concentration of 3% w/v showed a slow initial burst, and it sustained for up to 90 days (almost 100%). Antigen integrity was investigated by SDS-PAGE and the results indicated that TT released from the heparin-stabilized microspheres remains intact, when compared to the plain TT. The microspheres prepared without the stabilizer showed little or no band representing the protein aggregation or coagulation at the O/W interface. These results provide strong evidence that the protein stabilizer (heparin) played a role in preventing the inactivation of TT during microencapsulation (CitationCleland and Jones 1996).
For in vivo immunogenicity studies, we selected different combinations, as depicted in , and 3% w/v of heparin-stabilized TT-encapsulated microspheres were used as CHBMS. The results were compared with those of CRITT. A total of six groups or combinations were used, and the maximum antibody level of 4.5 IU/mL was observed with combination-1 after one year following the second booster dose. The antibody level attained in this study was a little higher than in the previous studies (CitationArthanari et al. 2011, Citation2014, CitationJaganathan et al. 2005). The results of in vivo studies indicated that CHBMS have potential application in the field of vaccine delivery and could be a more appropriate and better choice for the development of single-shot vaccine against tetanus.
Conclusion
From the results of our study, we have concluded that the 8/2 ratio of chitosan/HPMC can be used to prepare smooth, spherical microspheres, and a heparin concentration of 3% w/v is suitable for better in vitro release characteristics. Moreover, the potency of the TT-encapsulated CHBMS was tremendously increased after 1 year with the second booster dose, when compared with the potency of CRITT. Finally, we have concluded that chitosan–HPMC microspheres will be a good alternative adjuvant for the development of single-dose vaccine in the near future.
Acknowledgements
The authors acknowledge Dr. Sourabh Sharma and Dr. Tejpal Kashyap, DTP section, Central Research Institute, Kasauli, India for providing the gift sample of plain tetanus toxoid.
Declaration of interest
The authors have no declaration of interest and all the authors are responsible for the entire content of this paper.
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (Grant No. 2014-004694).
References
- Alpar HO, Almeida AJ, Brown MRW, Williamson ED. 1994. Immune responses to mucosally administered tetanus toxoid in biodegradable PLA microspheres. Proc Int Cont Rel Bioact Mater. 21:867–868.
- Arthanari S, Renukadevi P, Mani KR. 2011. Preparation and evaluation of sucrose stabilized tetanus toxoid encapsulated into chitosan microspheres. Gen Med Biomark Health Sci. 3:91–97.
- Arthanari S, Renukadevi P, Saravanakumar V. 2014. Evaluation of lactose stabilized tetanus toxoid encapsulated into alginate, HPMC composite microspheres. J Ind Eng Chem. 20:2018–2022.
- Barman SP, Lunsford L, Chambers P, Hedley ML. 2000. Two methods for quantifying DNA extracted from poly (lactide-co-glycolide) microspheres. J Control Release. 69:337–344.
- Bilati U, Allemann E, Doelker E. 2005. Strategic approaches for overcoming peptide and protein instability within biodegradable nano- and microparticles. Eur J Pharm Biopharm. 59:375–388.
- Cleland JL, Barron L, Berman PW, Daugherty A, Gregory T, Lim A. 1996. Development of a single shot sub unit vaccine for HIV-1 and 2. Defining optimal auto boost characteristics the humoral immune response. J Pharm Sci. 85:1346–1349.
- Cleland JL, Jones AJS. 1996. Stable formulations of recombinant human growth hormone and interferon-gamma for microencapsulation in biodegradable microspheres. Pharm Res. 13:1464–1475.
- Cox JC, Coulter AR. 1997. Adjuvants – a classification and review of their modes of action. Vaccine. 15:248–256.
- Derek T, O’Hagan DT, Rappuoli R. 2004. Novel approaches to vaccine delivery. Pharma Res. 21:1519–1530.
- Eldridge JH, Gilley RM, Staas JK, Moldoveanu A, Merlbroek JA, Tice TR. 1989. Biodegradable microspheres: a vaccine delivery system for oral immunization. Curr Top Microbial Immun. 146:59–66.
- Eldridge JH, Staas JK, Meulbroek JA, Tice TR, Gilley RM. 1991. Biodegradable and biocompatible poly (dl-lactide co-glycolide) microspheres as an adjuvant for staphylococcal enterotoxin B toxoid which enhances the level of toxin-neutralizing antibodies. Infect Immun. 59:2978–2986.
- Felt O, Buri P, Gurny R. 1998. Chitosan: a unique polysaccharide for drug delivery. Drug Dev Ind Pharm. 24:979–993.
- Gupta RK, Siber GR. 1995. Adjuvants for human vaccines – current status, problems and future prospects. Vaccine. 13:1263–1276.
- Hirrien M, Chevillard C, Desbrieres J, Axelos MAV, Rinaudo M. 1998. Thermogelation of methylcelluloses. New evidence for understanding the gelation mechanism. Polymer. 39:6251–6259.
- Hirrien M, Chevillard C, Desbrieres J, Axelos MAV, Rinaudo M. 1996. Physical properties of methylcelluloses in relation with the conditions for cellulose modification. Carbohyd Poly. 31:243–252.
- Ibbett RN, Philp K, Price DM. 1992. 13C n.m.r. studies of the thermal behaviour of aqueous solutions of cellulose ethers. Polymer. 19:4087–4094.
- Illum L. 1998. Chitosan and its use as a pharmaceutical excipient. Pharm Res. 15:1326–1331.
- Indian Pharmacopoeia. 1985. Ministry of Health and Family Welfare. Government of India, 2: A39.
- Jaganathan KS, Rao YUB, Singh P, Prabakaran D, Gupta S, Jain A, Vyas SP. 2005. Development of a single dose tetanus toxoid formulation based on polymeric microspheres: a comparative study of poly (d,l-lactic-co-glycolic acid) versus chitosan microspheres. Int J Pharm. 294:23–32.
- Jameela SR, Mishra A, Jayakrishnan A. 1994. Cross-linked chitosan microspheres as carriers for prolonged delivery of macromolecular drugs. J Biomat Sci Polym Edu. 6:621–632.
- Joane RJ, Frier M, Perkins AC, Jones NS, Davis SS, Illum L. 1999. Evaluation of the clearance characteristics of bio adhesive systems in humans. Int J Pharm. 178:55–65.
- Johansen P, Men Y, Audran R, Corradin G, Merkle HP, Gander B. 1998. Improving stability and release kinetics of microencapsulated tetanus toxoid by co-encapsulation of additives. Pharm Res. 15:1103–1110.
- Kohn J, Niemi SM, Albert EC, Murphy JC, Langer RS, Fox JG. 1986. Single step immunization using a controlled release, biodegradable polymer with sustained adjuvant activity. J Immunol Met. 95:31–38.
- Kreuter J, Speiser P. 1976. New adjuvants on a polymethyl methacrylate base. Infect Immun. 13:204.
- Lutsiak CME, Kwon GS, Samuel J. 2006. Biodegradable nanoparticle delivery of a Th2-biased peptide for induction of Th1 immune responses. J Pharm Pharmacol. 58:739–747.
- O’Hagan DT, Jeffery H, Davis SS. 1993. Long term antibody responses in mice following subcutaneous immunization with ovalbumin entrapped in biodegradable microspheres. Vaccine. 11:965–969.
- O’Hagan DT, Rahman D, McGee JP, Jeffery H, Davies MC, Williams P. 1991. Biodegradable microparticles as controlled release antigen delivery system. Immunology. 73:239–242.
- Oladiran I, Meier DE, Ojelade AA, OlaOlarun DA, Adeniran A, Tarpley JL. 2002. Tetanus: continuing problem in the developing world. World J Surg. 26:1282–1285.
- Petrovsky N, Aguilar JC. 2004. Vaccine adjuvants: current state and future trends. Immunol Cell Biol 82:482–496.
- Singh M, Li XM, Wang H, McGee JP, Zamb T, Koff W, et al. 1997. Immunogenicity and protection in small-animal models with controlled-release tetanus toxoid microparticles as a single-dose vaccine. Infect Immun. 65:1716–1721.
- Trujillo Vargas CM, Mayer KD, Bickert T, Palmetshofer A, Grunewald S, Ramirez Pineda JR, et al. 2005. Vaccinations with T helper type 1 directing adjuvants have different suppressive effects on the development of allergen-induced T helper type 2 responses. Clin Exp Allergy. 35:1003–1013.
- Vandelaer J, Birmingham M, Gasse F, Kurian M, Shaw C, Garnier S. 2003. Tetanus in developing countries; an update on the Maternal and Neonatal Tetanus Elimination Initiative. Vaccine. 21: 3442–3445.
- van der Lubben LM, Verhoef JC, Borchard G, Junginger HE. 2001. Chitosan for mucosal vaccination. Adv Drug Deliv Rev. 52:139–144.
- Warren HS, Vogel FR, Chedid LA. 1986. Current status of immunological adjuvants. Ann Rev Immunol. 4:369–376.
- Zhao Z, Leong KW. 1996. Controlled delivery of antigens and adjuvants in vaccine development. J Pharm Sci. 85:1261–1270.