
Abstract
The aim of this study was to develop novel biomedicated electrospun nanofibers for controlled release. Pre-formulation studies were carried out for nanofibers of sodium alginate (SA) (2 wt %)/polyvinyl alcohol (PVA) (10 wt %) composites (2/8, 3/7 and 4/6), by an electrospinning technique. The morphology and average diameter of the nanofibers were investigated by scanning electron microscopy (SEM). The optimum ratio (3/7) was used to load gatifloxacin hydrochloride (GH) (1wt %), found to form smooth fibers with uniform structures. The drug entrapment in the composite nanofibers was confirmed by SEM, X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), thermo gravimetric analysis (TGA), differential scanning calorimetry (DSC), and swelling behavior. The drug release behavior was investigated using phosphate-buffered saline (PBS) (pH 7.4) at 37°C for 24 h. The XRD and FTIR data demonstrate that there are good interactions between PVA and SA, possibly caused by hydrogen bonds. As much as 90% of the GH was released from the electrospun fibers within 6 h of incubation. Beyond this, the release was sustained for 24 h. The thickness of nanofibers greatly influenced the initial release and rate of drug release. Moreover, GH-loaded sodium alginate/PVA composite nanofibers exhibited a useful and convenient method for electrospinning in order to control the rate and period of drug release in wound-healing applications.
Introduction
Over the last decade, extensive studies have been conducted to investigate drug delivery systems, to improve the therapeutic effect and to reduce the toxicity of conventional dosage forms. Recently, the nanoscale formulations such as liposomes, polymeric micelles and nanofibers have attracted special attention, to develop controlled drug release. Electrospun nanofibers offer great flexibility in the selection of materials or drugs for drug delivery applications, compared with other types of formulations. In addition, the nanofibers have high loading capacity, high encapsulation efficiency, and provide simultaneous delivery of diverse therapies, ease of operation and cost effectiveness, which are promising features for use in drug delivery (CitationHu et al. 2014).
There are different materials, including natural polymers and synthetic polymers, and hybrid blends of the two have been used to prepare electrospun fibers. Due to the low hydrophilicity of synthetic polymers, natural polymers have recently been used. Natural polymers exhibit better biocompatibility and low immunogenicity, and some even exhibit antibacterial property. Various studies have reported or summarized that natural polymers, and their derivatives and composites, are used to prepare electrospun fibers (CitationBhardwaj and Kundu 2010, CitationSchiffman and Schauer 2008, CitationWu et al. 2009). One of the previous reviews summarized that the characteristics of various electrospinning techniques using polysaccharides such as alginate, cellulose, chitin, chitosan, hyaluronic acid, starch, dextran, and heparin, have been applied in the biomedical field for tissue engineering, wound dressing, drug delivery and enzyme immobilization (CitationLee et al. 2009).
Alginate (SA), a natural polymer composed of (1, 4)-linked β–D-mannuronic acid and α– L-guluronic acid, has been extensively studied for a wide variety of biological applications, due to its low cost, biocompatibility, biodegradability, and ease of chemical derivatization (CitationStone et al. 2013). Alginate-based beads demonstrate pH sensitivity and stability, and have been developed for controlled delivery systems (CitationBadwan et al. 1985). Due to its high rigidity and fragility, alginate is usually used as a co-polymer, and is blended with a compatible flexible vinyl polymer (CitationCaykara and Demirci 2006). Although the natural polymers have good biocompatibility and biodegradability, they have low mechanical properties and some processing difficulties, compared with synthetic polymers. Few reports have validated this capacity of alginate to be electrospun with synthetic polymers like composite nanofibers of poly vinyl alcohol (PVA) and poly (ethylene oxide) (PEO) (CitationShalumon et al. 2011).
PVA is a biodegradable synthetic polymer possessing appropriate mechanical properties, and is biocompatible, nontoxic, and noncarcinogenic (CitationPal et al. 2006, CitationYu et al. 2006). Owing to these properties, PVA is used in the biomedical field, in products such as soft contact lenses, implants, cartilage, skin, and cardiovascular devices (CitationYu et al. 2006, CitationKenawy et al. 2007). To overcome the poor stability of PVA in water, it has been made insoluble by grafting, copolymerizing, and cross-linking. For wound-dressing, PVA has been used by cross-linking with polyvinyl pyrrolidine (PVP), SA, and sterculia, to provide a very quick burst and sustained release for 90, 5, and 24 h respectively (CitationJannesari et al. 2011).
Gatifloxacin, a third generation fluoroquinolone antibiotic, has broad spectrum activity, and is especially potent against gram positive bacteria including penicillin- resistant Streptococcus pneumoniae, and shows excellent activity against gram negative bacteria and atypical organisms as well (CitationFish and North 2001). In the present investigation, hydrophilic PVA–SA composite nanofibers are developed by electrospinning, and the drug gatifloxacin is loaded into the composite nanofibers by active loading (PVA–SA–GH). The PVA–SA–GH nanofibers formed are investigated by release characteristics through in vitro studies.
Materials and methods
Gatifloxacin, SA (medium viscosity), and PVA (Mowiol® 20–98) (molecular weight 125 000) were purchased from Sigma (Sigma-Aldrich, St Louis, MO). All other chemicals used in this study were of analytical grade.
Pre-formulation studies
The SA (2 wt %) and PVA (10 wt %) were prepared separately, with PVA being prepared by heating at 90°C for 1 to 2 h. Then the SA and PVA were mixed at various ratios (3/7, 2/8, and 1/9) (CitationShalumon et al. 2011). First, the solutions of PVA and SA were mixed by magnetic stirring overnight. Then, the PVA–SA blended solution was transferred to a plastic syringe equipped with a flat-end metal needle with an inner diameter of 0.7 mm. A voltage of 20 kV was supplied, with the feed rate of blended solution kept at 0.1 ml/h. The collection plate of aluminum foil was located at a fixed distance of 10 cm from the needle tip. The PVA–SA nanofibers were carefully removed from the collector and dried under vacuum at room temperature, for the complete removal of solvent.
Preparation of PVA–SA–GH nanofibers
The PVA–SA–GH nanofibers were prepared by the same method as used for PVA-SA nanofiber preparation, but to ensure good dispersion of the gatifloxacin in the blended solution, the gatifloxacin (1% w/v) was dissolved in PVA by magnetic stirring for 3 h. Then, the SA was mixed into the solution and the magnetic stirring was continued overnight.
Characterization of PVA–SA–GH nanofibers
The diameter and morphology of the PVA–SA–GH electrospun nanofibers were observed using a scanning electron microscope (SEM, JEOL JSM 5600, Tokyo, Japan). Each sample was sputter-coated with gold prior to analysis, and the nanofibers were analyzed in different positions.
To know the structural properties of gatifloxacin, PVA–SA, and PVA–SA–GH composite nanofibers, the XRD analysis (XRD, Rigaku, Tokyo, Japan) was carried out, at diffraction angles ranging between 10° and 50°.
The gatifloxacin, PVA–SA, and PVA–SA–GH composite nanofibers were characterized by FTIR (Fourier-transform infrared spectroscopy) (Nicolet-200, Thermo, USA), using KBr pellets.
Differential scanning calorimetry (DSC, Scinco N-650, Seoul, Korea) was performed separately for gatifloxacin, PVA–SA and PVA–SA–GH composite nanofibers. The samples were placed in crimped aluminum pans and heated from 0°C to 400°C, at a heating rate of 10°C per minute.
The stability of gatifloxacin, PVA–SA, and PVA–SA–GH composite nanofibers were studied using a thermo-gravimetric analyzer (TGA, Scinco Co. Ltd, Korea), in air atmosphere at 10°C/min.
Drug entrapment efficiency
The drug entrapment efficiency of nanofibers was calculated by drying the drug-loaded nanofibers at 40° C inside a hot air oven, and then the nanofiber mat of a known area (1 X 1 cm) was removed and dissolved in its respective solvent (water, in the case of PVA/SA nanofibers). The amount of drug in the respective solutions was calculated by UV analysis of these solutions, and was compared with the amount of drug that was loaded during the process of electrospinning of these fibers (CitationKataria et al. 2014).
Degree of swelling
The test was carried out separately for PVA–SA and PVA– SA–GH nanofibers in phosphate- buffered saline (PBS) of pH 7.4, at 37°C, for different time periods of 2, 4, 6, 8, 10 and 12 h. The degree of swelling of the drug-loaded nanofibers was calculated by the following equation (CitationKataria et al. 2014)
Where M is the weight of swollen nanofiber sample which was wiped dry with filter paper, and Md is the dried mass of sample immersed in buffer medium. It was measured by drying the swollen nanofibers in an oven at 40°C to get a constant weight.
In vitro drug release studies
To measure the drug release from the PVA–SA–GH electrospun nanofiber mats, a known area (1 × 1 cm) of drug-loaded nanofibers was placed into 100 ml of PBS (pH 7.4), and the release studies were carried out at 37°C, at 100 rpm, in a thermostatic shaking incubator. Samples of 5 ml were taken from the buffer solution after 0, 1, 2, 3, 4, 5, 6, 7, 8, 10, 14, 18, 21 and 24 h. After sampling, the volume removed was replaced with fresh medium. The amount of drug present in the release buffer was determined using a UV-Vis spectrophotometer at a λmax of 272 nm. The release experiments for each sample were performed in triplicate, and average values have been reported. The cumulative percentage drug release was calculated using the calibration curve of the drug in PBS (pH 7.4).
Results and discussion
Morphology of the nanofiber batch
The formation of hydrogen bond with water as well as with each polymer is due to the free solubility of PVA and SA in water. The morphologies of nanofibers with three different ratios of empty PVA/SA have been depicted in . For all nanofibers prepared, 2 wt % SA and 10 wt % PVA were used. The PVA–SA composite nanofibers, electrospun from 3/7 (SA/PVA) () exhibit smooth surface and uniform diameter, and the nanofibers prepared with ratios of 2/8 () and 1/9 () demonstrated very smooth clumping, which indicates lower encapsulation efficiency. To obtain fine, uniform fibers, the ratio of SA should be as high as possible for hemostasis, high entrapment efficiency, and wound healing properties (CitationFu et al. 2014). The optimum ratio found in our study was 3/7, of 2 wt % SA and 10 wt % PVA. reveals that gatifloxacin-loaded composite nanofibers (PVA – SA – GH), with a SA/PVA ratio of 3/7 yielded the most uniform fibrous morphology with smooth surface.
Figure 1. SEM micrograph of (A) 3/7, (B) 2/8, (C) 1/9 plain SA–PVA nanofibers, and (D) gatifloxacin-loaded PVA–SA nanofibers.

Characterization of drug-loaded electrospun nanofibers
Gatifloxacin, PVA–SA and PVA–SA–GH composite nanofibers were characterized using Fourier-transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), and Differential scanning calorimetry (DSC).
FTIR analysis
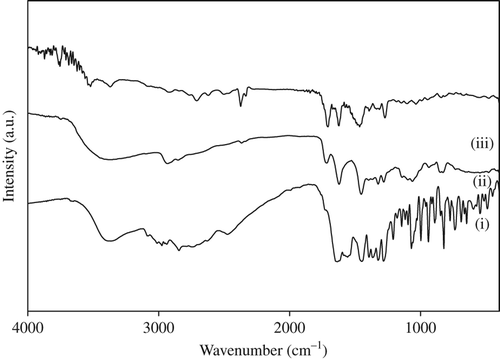
The functional groups and the bonding between polymer and drug were confirmed by characterizing the drug-loaded nanofiber patch by FTIR. The FTIR curve is shown in , which represents (i) gatifloxacin, (ii) PVA–SA composite nanofiber, and (iii) drug-loaded PVA–SA composite nanofiber. In (curve i), the FTIR spectrum of gatifloxacin and its characteristic peaks were assigned as follows: 3366 cm− 1 (O–H group, H-bonded), 2975, 2844 cm−1 (C–H group, stretching), 1635 cm− 1 (C = O group, stretching), 1449 cm− 1 (C = C group, stretching), 1393, 1365, and 1323 cm− 1 (C–F group, stretching). These characteristic adsorption peaks were absent in the FTIR spectrum of PVA–SA–GH composite nanofibers, suggesting the interaction of GH with polymer materials. The FTIR spectrum of PVA–SA composite nanofibers (curve ii, ) consists of the following peaks: broad peak at 3375 cm− 1 (O–H group, H-bonded), 2932 cm− 1 (C–H group, stretching), 1715 cm− 1 (C = O group, stretching). The broad peak of the hydroxyl group at 3375 cm− 1 is because of the hydrogen bonding between the hydroxyl groups of PVA and SA. The band between 3000 and 2900 cm− 1 represents alkane and aromatic C–H stretching. The peak between 1650 and 1200 cm− 1 indicates the presence of double-bonded functional groups. In the spectrum for PVA–SA–GH composite nanofibers (curve iii, ), the following peaks were observed: the peaks between 1650 cm− 1 and 1600 cm− 1 were assigned to quinolones, and those between 3700 and 3400 cm− 1 were assigned to hydroxyl (O–H) groups. The peaks between 1300 and 1200 cm− 1 suggested bending vibration of O–H groups, which proved the presence of carboxylic acid.
Figure 2. FTIR spectra of (i) gatifloxacin, (ii) PVA–SA composite nanofibers, and (iii) drug loaded PVA–SA nanofibers.
TGA analysis
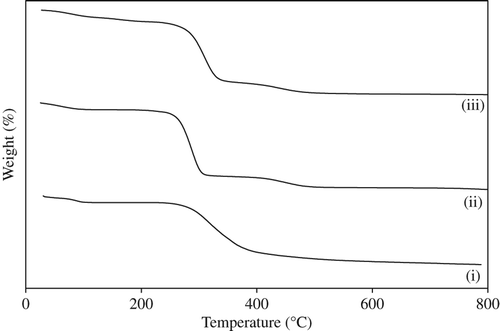
shows the TGA analysis of (i) gatifloxacin, (ii) PVA–SA composite nanofibers, and (iii) drug-loaded PVA–SA composite nanofibers, which shows the thermal stability of prepared nanofibers before and after drug loading. For all samples, two degradation points were observed. One weight loss occurs at 20–100°C, which indicates the moisture vaporization, independent of the composition of all samples. The second weight loss, at 250–300°C, is due to the thermal degradation of the prepared nanofibers, and it continues up to 800°C.
Figure 3. TGA of (i) gatifloxacin, (ii) PVA–SA composite nanofibers, and (iii) drug-loaded PVA–SA nanofibers.
XRD analysis
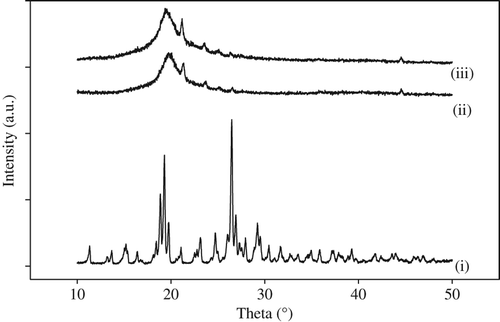
The XRD spectra of (i) gatifloxacin, (ii) PVA–SA composite nanofibers, and (iii) PVA–SA–GH composite nanofibers are shown in . In the PVA–SA composite nanofibers, the characteristic peak was observed for semi-crystalline PVA at 19.74° (curve ii), which is due to the occurrence of strong intermolecular and intramolecular hydrogen bonding (CitationZhang et al. 2007). There were three sharp peaks at around 2θ = 19.22, 19.3 and 26.48 observed, due to the crystalline character of the drug (curve i). In the drug-loaded PVA–SA composite nanofibers, the crystalline nature of the drug changed due to interaction and the entrapment of the drug into polymer composites. It was confirmed that the peak was shifted from 2θ = 19.74° to a lower diffraction angle (2θ = 19.36°) (curve iii). Finally, it was confirmed that the drug was entrapped into polymer composites.
Figure 4. XRD of (i) gatifloxacin, (ii) PVA–SA composite nanofibers, and (iii) drug-loaded PVA–SA nanofibers.
DSC analysis
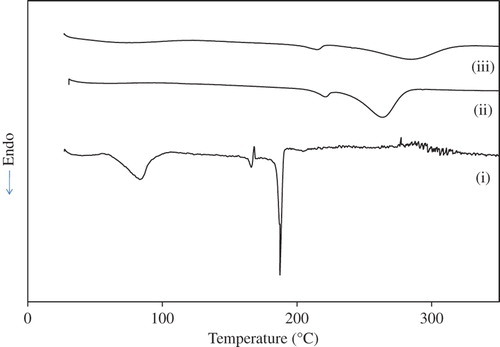
The DSC thermograph of (i) gatifloxacin, (ii) PVA–SA composite nanofiber, and (iii) PVA–SA– GH composite nanofiber is shown in . In PVA–SA composite nanofiber (curve ii), the endothermic peaks at 220°C and 263°C correspond to the melting and decomposition temperatures, respectively. Moreover, the characteristic peaks of gatifloxacin (curve i) are absent in the drug-loaded PVA–SA composite nanofiber (curve iii), indicating the amorphous dispersion of drug into the PVA–SA composites. However, in the case of drug-loaded PVA–SA composite nanofibers, the decomposition occurs at high temperature (285°C) as compared with the drug and PVA–SA composite nanofiber. This clearly demonstrates that the stability of the drug was enhanced due to its encapsulation between polymeric chains.
Figure 5. DSC pattern of (i) gatifloxacin, (ii) PVA–SA composite nanofibers, and (iii) drug-loaded PVA–SA nanofibers.
Drug entrapment efficiency
The efficiency of entrapment of the drug into nanofibers depends on the solubility of drug in the composite polymer solution. Due to the nonvolatile nature of the drug, the entrapment efficiency calculated by UV spectrophotometry was found to be 95%.
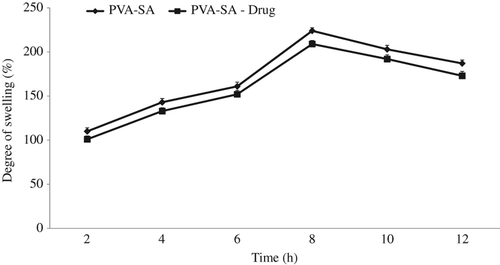
Degree of swelling
The degree of swelling of nanofibers plays an important role in the loading and release behavior of the drug. shows the degree of swelling of PVA–SA composite nanofibers and PVA–SA– GH composite nanofibers at different time intervals. The swelling pattern was observed for plain and drug-loaded nanofibers, and it was found to be 110 ± 4% and 101 ± 3.8% for 2 h 43 ± 4.2% and 133 ± 3.7% for 4 h, 161 ± 4.8% and 152 ± 4.5% for 6 h, 224 ± 3.5% and 209 ± 4.1% for 8 h, 203 ± 4.6% and 192 ± 4.8% for 10 h, 187 ± 3.9% and 173 ± 4.9% for 12 h respectively. From the results, it was clearly demonstrated that the degree of swelling of drug-loaded nanofibers was decreased due to the water solubility of the drug, which causes interaction of the polymeric chain that restricts the swelling. However, in all cases, the maximum swelling was attained at 8 h, beyond which the degree of swelling was decreased. The reason for this behavior (decreased swelling) may be due to the degradation and leaching of polymeric composites after 8 h (CitationKataria et al. 2014).
Figure 6. Degree of swelling of PVA–SA composite nanofibers and drug-loaded PVA–SA nanofibers.
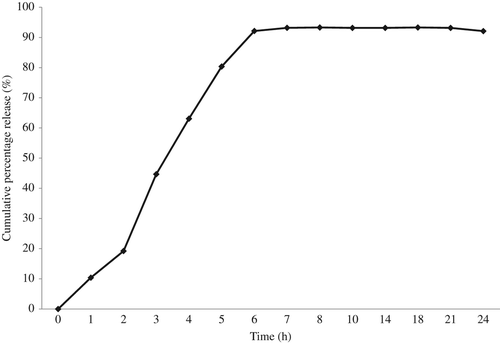
In vitro drug release
shows the in vitro drug release pattern of the PVA– SA– GH nanofibers, and the in vitro release profile specifies that approximately 95% cumulative percentage release was observed after 8 h. The drug was released with a very slight burst initially (10% in 1 h) and the burst effect was tremendously increased within 3 h (44%). The maximum drug was released in a sustained manner around a period of 8 h (93%), beyond which no further release was observed. From the release pattern, it was summarized that the diffusion-based release occurred for the initial period of 1–2 h, beyond which it was based on the erosion of the polymers until the 8th hour, when the maximum drug was released.
Figure 7. Drug release pattern of drug-loaded PVA–SA nanofibers.
Conclusion
Gatifloxacin-loaded PVA–SA composite nanofibers were developed by the electrospinning technique. Pre-formulation studies were performed with nanofibers at ratios of plain SA/PVA of 2/8, 3/7 and 4/6, and the ratio of 3/7 was found to yield uniformly sized nanofibers with smooth surface. The drug was loaded by active loading, and the entrapment efficiency was 95%. Different characterization studies were performed, which confirmed the drug entrapment, and the stability of drug-loaded nanofibers. Due to the water solubility of the drug, the degree of swelling was decreased in the drug-loaded PVA–SA composite nanofibers. The in vitro release profile shows controlled continuous release of the drug within 6 h, from the gatifloxacin loaded PVA–SA composite nanofibers. Finally, it can be concluded that the drug loaded PVA–SA composite nanofibers would be a good choice for a drug delivery system, without any side effects.
Acknowledgement
The authors are highly grateful to the Seoryeong High School, Seosan-Si, South Korea, for their financial support.
Declaration of interest
The authors have no declaration of interest, and all the authors are responsible for the entire content of this paper.
References
- Badwan AA, Abumalooh A, Sallam E, Abukalaf A, Jawan OA. 1985. Sustained release drug delivery systems using calcium alginate beads. Drug Dev Ind Pharm. 11:239–256.
- Bhardwaj N, Kundu SC. 2010. Electrospinning: a fascinating fiber fabrication technique, Biotechnol Adv. 28:325–347.
- Caykara T, Demirci S. 2006. Preparation and characterization of blend films of poly (vinyl alcohol) and sodium alginate. J Macromol Sci A. 43:1113–1121.
- Fish DN, North DS. 2001. Gatifloxacin, an advanced 8-methoxy fluroquinolone. Pharmacother. 21:35–59.
- Fu R, Li C, Yu C, Xie H, Shi S, Li Z, et al. 2014. A novel electrospun membrane based on moxifloxacin hydrochloride/poly(vinyl alcohol)/sodium alginate for antibacterial wound dressings in practical application. Drug Deliv. (doi:10.3109/10717544.2014.918676)
- Hu X, Liu S, Zhou G, Huang Y, Xie Z, Jing X. 2014. Electrospinning of polymeric nanofibers for drug delivery applications. J Control Release. 185:12–21.
- Jannesari M, Varshosaz J, Morshed M, Zamani M. 2011. Composite poly (vinyl alcohol)/poly (vinyl acetate) electrospun nanofibrous mats as a novel wound dressing matrix for controlled release of drugs. Int J Nanomed. 6:993–1003.
- Kataria K, Gupta A, Rath G, Mathur RB, Dhakate SR. 2014. In vivo wound healing performance of drug loaded electrospun composite nanofibers transdermal patch. Int J Pharm. 469:102–110.
- Kenawy ER, Hay EA, Newehy MHE, Wnek GE. 2007. Controlled release of ketoprofen from electrospun poly (vinyl alcohol) nanofibers. Mater Sci Eng A. 459:390–396.
- Lee KY, Jeong L, Kang YO, Lee SJ, Park WH. 2009. Electrospinning of polysaccharides for regenerative medicine, Adv Drug Deliv Rev. 61:1020–1032.
- Pal K, Banthia AK, Majumdar DK. 2006. Polyvinyl alcohol-glycine composite membranes: preparation, characterization, drug release and cytocompatibility studies. Biomed Mater. 1:49–55.
- Schiffman JD, Schauer CL. 2008. A review: electrospinning of biopolymer nanofibers and their applications Polym Rev. 48:317–352.
- Shalumon KT, Anulekha KH, Nair SV, Chennazhi KP, Jayakumar R. 2011. Sodium alginate/poly (vinyl alcohol)/nano ZnO composite nanofibers for antibacterial wound dressings. Int J Biol Macromol. 49:247–254.
- Stone SA, Gosavi P, Athauda TJ, Ozer RR. 2013. In situ citric acid crosslinking of alginate/poly vinyl alcohol electrospun nanofibers. Mater Lett. 112: 32–35.
- Wu Y, MacKay JA, McDaniel JR, Chilkoti A, Clark RL. 2009. Fabrication of elastin-like polypeptide nanoparticles for drug delivery by electrospraying. Biomacromolecules. 10:19–24.
- Yu H, Xu H, Chen X, Hao J, Jing X. 2006. Medicated wound dressing on poly (vinyl alcohol)/poly(N-vinyl pyrrolidone)/chitosan hydrogels. J Appl Polym Sci. 101:2453–2463.
- Zhang Y, Huang X, Duan B, Wu L, Li S, Yuan X. 2007. Preparation of electrospun chitosan/poly (vinyl alcohol) membranes. Colloid Polym Sci. 285:855–863.