
Abstract
In spite of specific immune effector mechanisms raised against tumor cells, there are mechanisms employed by the tumor cells to keep them away from immune recognition and elimination; some of these mechanisms have been identified, while others are still poorly understood. Manipulation or augmentation of specific antitumor immune responses are now the preferred approaches for treatment of malignancies, and traditional therapeutic approaches are being replaced by the use of agents which potentiate immune effector mechanisms, broadly called “immunotherapy”. Cancer immunotherapy is generally classified into two main classes including active and passive methods. Interventions to augment the immune system of the patient, for example, vaccination or adjuvant therapy, actively promote antitumor effector mechanisms to improve cancer elimination. On the other hand, administration of specific monoclonal antibodies (mAbs) against different tumor antigens and adoptive transfer of genetically-modified specific T cells are currently the most rapidly developing approaches for cancer targeted therapy. In this review, we will discuss the different modalities for active and passive immunotherapy for cancer.
Historical background
In 1909, the Nobel Prize winner Paul Ehrlich first proposed the concept that transformed cells continuously arise in our bodies, and elements of the immune system contribute to the control of these transformed cells and eradicate these malignant cells before they are manifested clinically (CitationKim et al. 2007, CitationEhrlich and Himmelweit 1956).
In the mid-20th century, Burnet and Thomas developed the immune surveillance hypothesis which postulated that the immune system very efficiently destroys malignant cells, and experimental results showing strong immune-mediated rejection of transplanted tumors in mice supported this idea. However, this hypothesis was challenged by experimental data and clinical observations in the 1970s, which indicated that immunosuppressive medication for organ transplantation did not increase the incidence of solid tumors in areas such as colon, lung and breast (CitationBurnet 1957, CitationNewstead 1998).
Although the immune system does not play the central role in the immune surveillance as suggested by Burnet and Thomas, more recent clinical studies provide evidence that immune effector cells and mediators such as B, T, natural killer (NK), natural killer T (NKT) cells, and cytokines contribute to the control of premalignant cells (CitationRiether et al. 2013).
Despite these effector mechanisms, malignant cells are able to evade immune responses. In this regard, immunotherapy for the treatment of cancer has been used for decades and has been dramatically developed during recent years. In this review, we provide a general overview of cancer-active immunotherapy [for example, tumor vaccination using tumor-derived peptide and protein, DNA and dendritic cell-based vaccine, and adjuvant therapy using BCG (Bacillus Calmette-Guérin), and cytokines] and passive immunotherapy [for example, using monoclonal antibodies (mAbs) and adoptive T cell therapy], as well as approaches involving the targeting of cancer stem cells (CSCs) to overcome cancer relapse and provide resistance.
Tumor escape from immune system
In spite of several mechanisms active in the immune system to recognize and eliminate tumor cells, known as the “elimination phase”, some variants of these cells selectively acquire increased resistance against immune responses (equilibrium phase). Thereafter, resistant cells continue to grow by employing mechanisms to evade the immune responses (escape phase). During this phase, tumor cells develop resistance against both innate and adaptive immune mechanisms and the clinical manifestations of tumor develop. All the three above-mentioned phases are collectively called “cancer immunoediting”, believed to be a phenomenon derived by immune responses (CitationDunn et al. 2002, CitationFinn 2012). In the escape phase, resistant tumor cells develop two classes of mechanisms to avoid being killed by what is known as “immunosurveillance”: (I) immunologic ignorance and tolerance, and (II) negative regulation of immune cells (CitationBhutia et al. 2010). The selection of tumor cells with weak or no expression of specific antigens, defects in the expression of MHC molecules and antigen-processing and presentation, and the presentation of tumor antigen-derived peptides with weak or no expression of costimulatory and adhesion molecules are mechanisms proposed to be developed by tumor cells than enable them to be tolerated or ignored (CitationCostello et al. 1999, CitationMaeurer et al. 1996, CitationIgney and Krammer 2002, CitationFlynn and Stockinger 2003). These mechanisms are not recently discovered, but the induction of regulatory mechanisms continues to be clarified. So far, expression of FasL to induce apoptosis in immune effector cells (a phenomenon called Fas counterattack) (CitationStrand and Galle 1998), expression of programmed death-ligand 1 (PD-L1) to inhibit immune effector mechanisms (CitationIwai et al. 2002), production of immunosuppressive cytokines, for example, transforming growth factor (TGF)-β (CitationPasche 2001), interleukin (IL)-10 (CitationSalazar-Onfray 1999, CitationMocellin et al. 2005), vascular endothelial growth factor (VEGF) (CitationGabrilovich et al. 1996), induction of T cell anergy via production of indoleamine 2,3-dioxygenase (IDO) and depletion of tryptophan (CitationMellor et al. 2003), and the release of exosomes or microvesicles by tumor cells, have been revealed (CitationWhiteside 2005, CitationTaylor et al. 1988, CitationKim et al. 2005).
Adjuvant therapy
The effect of an adjuvant in immunotherapy is in potentiating immune response against tumor cells. As discussed earlier, different approaches have been used to vaccinate cancer patients and raise specific immune responses for treatment. Parallel administration of an adjuvant could induce more potent and effective immune responses and therefore increase the efficacy of active immunotherapy. As cancer patients are generally immunocompromised and vaccination is usually done by self-derived antigens, adjuvants for therapy should be more potent than adjuvants for prophylaxis, but with acceptable toxicity and safety. Some agents have been used for this purpose (CitationMesa and Fernandez 2004).
Nowadays, monotherapy by cytokines and other immunological response-modifiers have been considered as immunotherapy by adjuvants or in “adjuvant therapy” for cancer. The best-defined example is the intravesical administration of BCG for treatment of bladder cancer. The exact mechanism of such treatment is not fully understood, but it has been proposed that the presence of bacteria in malignant tissue could induce inflammation and the recruitment of immune cells that eventually lead to the elimination of cancerous cells (CitationMeyer et al. 2002). Interferon (IFN)-α is currently used as the treatment regimen for melanoma (CitationHauschild 2009), AIDS-related Kaposi's sarcoma (CitationAbrams and Volberding 1986), renal cancer (CitationMinasian et al. 1993), hairy cell leukemia (HCL) (CitationAhmed and Rai 2003), and chronic myelogenous leukemia (CML) (CitationBonifazi et al. 2001). The overall mechanism of IFN-α is proposed to be increased expression of MHC I and antigen presentation for better and more effective antitumor immune responses (CitationKirkwood 2002). IL-2 has been used to reverse dysfunction of immune effector cells in the treatment of metastatic melanoma (CitationAtkins et al. 1999) and renal cell carcinoma (CitationDutcher 2011). Recently, stimulating innate immunity by the ligands of toll-like receptors (TLRs) or their analogs has been considered as a new category of cancer immunotherapeutics. Imiquimod activates TLR7 on immune cells, and is used as cream for treatment of basal cell carcinoma (BCC), the most common form of skin cancer (CitationOldfield et al. 2005). Resiquimod (R-848), on the other hand, stimulates TLR7 and TLR8 and is used to cure viral skin lesions and skin cancers (CitationMeyer et al. 2013).
Passive immunotherapy
Therapeutic monoclonal antibodies
More than 100 years ago, using “magic bullets” to kill tumor cells and treat cancers was proposed by Paul Erlich. This interesting idea was initially promising, but encountered some clinical and experimental obstacles, including unknown purity of antibody preparations and therefore induction of some unwanted reactions, and failure to recognize definite tumor targets (CitationOldham 1983, CitationScott et al. 2012). Several years later, the advent of mAb production technology developed by the tireless efforts and experiments of George Köhler and Cesar Milstein in 1975 (CitationKohler and Milstein 1975), solved the problems. Subsequently, a vast number of mAbs was produced against different known targets for diverse clinical and diagnostic proposes. The first Food and Drug Administration (FDA)-approved therapeutic mAb was anti-CD3 mouse mAb, so-called OKT3, to prevent rejection of kidney allograft (CitationGroup 1985). The murine origin of OKT3, production of human anti-mouse antibody (HAMA) in humans, and also improper interaction of mouse mAb with human effector molecules, that is, C1 and FcγRs, greatly limited the applications of mouse mAbs in chronic clinical administrations (CitationBaker et al. 2010). As shown in , only two other FDA-approved mouse mAbs cancer therapeutics are designed for clinical settings, as conjugated platforms to deliver the radioisotope payload to the tumor mass. By exchanging the constant (c) regions of mouse mAb for human ones (), “chimeric” mAb was introduced with better biologic effects and lower immunogenicity in humans (CitationNeuberger et al. 1985). FDA-approved chimeric mAbs for cancer therapy have been listed in . As antibody specificity is determined by the complementarity-determining regions (CDRs) of variable (V) domains of mAb, but not the whole V domain, CDRs from mouse mAb were “grafted” onto the backbone of the human Ig molecule to produce the third generation of therapeutic mAbs, called “humanized” mAbs (CitationJones et al. 1986) (). FDA-approved humanized mAbs for cancer therapy have also been listed in . However, because the process of producing humanized mAb with better efficacy consumed too much time and effort, and the immunogenicity of the end product was too low, a fourth generation of therapeutic mAbs were produced using advanced in vitro and in vivo methods such as phage display (CitationSmith 1985), the Epstein–Barr virus (EBV)-transformation of B cells (CitationBorrebaeck 1989), and the use of transgenic mice (CitationLonberg 2008). Fourth generation mAbs are “fully-human” with any part of mouse origin. It is believed that these antibodies are not immunogenic, but allotypic differences between therapeutic mAb and recipient patient, and also idiotypic determinants residing in the antigen-binding site of therapeutic mAb can induce production of anti-antibodies and lead to neutralization of the drug and some clinically adverse effects (CitationHarding et al. 2010).
Figure 1. Generation of therapeutic monoclonal antibodies.
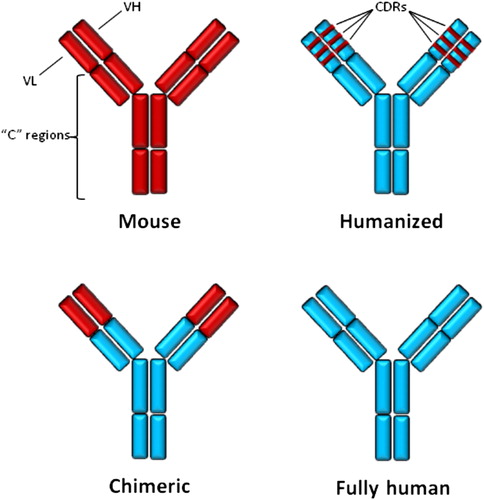
Table I. FDA-approved monoclonal antibodies for cancer therapy.
The mechanisms of action of therapeutic mAbs to induce anti-tumor effects have been explored exclusively with respect to antibody-dependent cell-mediated cytotoxicity (ADCC) for Herceptin (CitationLewis et al. 1993), Rituximab (CitationReff et al. 1994), and Ofatumumab (CitationTeeling et al. 2004); complement-dependent cytotoxicity (CDC) for Rituximab (CitationReff et al. 1994), Alemtuzumab (CitationZent et al. 2008), and Ofatumumab (CitationTeeling et al. 2004); interaction with growth factor receptors and inhibition of survival signaling for Herceptin (CitationLewis et al. 1993), Cetuximab (CitationHuang et al. 1999) and Panitumumab (CitationYang et al. 1999); antiangiogenic effect for Bevacizumab (CitationKim et al. 1992); prevention of bone destruction due to breast and prostate cancers for Denosumab (CitationPageau 2009, CitationSmith et al. 2012); and delivery of cytotoxic agents, for example, calicheamicin, yttrium-90, iodine-131, and monomethyl auristatin E to tumor cells for Gemtuzumab (CitationBreccia and Lo-Coco 2011) Ibritumomab (CitationJacobs 2007), Tositumomab (CitationRutar et al. 2001) and Brentuximab (CitationVaklavas and Forero-Torres 2012), respectively. It is noteworthy that each therapeutic mAb could also have its own specific function according to the nature of the targeted antigen. For example, Herceptin recognizes and binds to HER2 molecules and leads to internalization and inhibition of auto-cleavage of HER2. On the other hand, dimerization of HER2 to other members of the HER family could be inhibited by Herceptin (CitationVu and Claret 2012). Additionally, therapeutic mAb against CTLA-4 (Ipilimumab) antagonizes CD80 and CD86 molecules on antigen-presenting cells (APCs), and as a result, hinders inactivation of tumor-specific effector T cells (CitationTarhini et al. 2010).
New approaches for interfering with inhibitory pathways of the immune system to obtain prolonged antitumor cellular responses have been developed. Blocking or antagonistic mAbs against T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), lymphocyte activating gene 3 (LAG-3), B and T lymphocyte attenuator (BTLA), and programmed death-1 (PD-1) and its ligand PD-L1 have led to promising antitumor effects (CitationPardoll 2012, Kyi and Postow 2013). On the other hand, some experiments have been carried out to potentiate antitumor responses by administering agonistic mAbs against costimulatory and activatory molecules on T lymphocytes, for example, CD137, OX-40, 4-1BB (CD137), GITR, and CD27 (CitationMittler et al. 2004, CitationMoran et al. 2013, CitationSchaer et al. 2012, CitationVinay and Kwon 2012, CitationRiether et al. 2012).
Adoptive T cell therapy
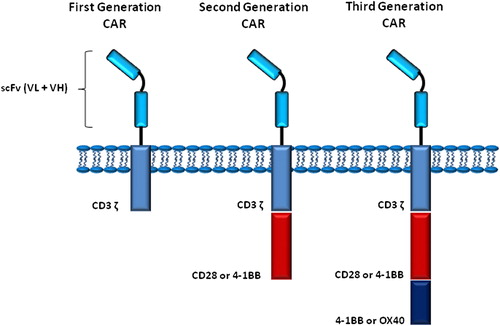
Adoptive transfer of ex vivo-enriched and expanded tumor-specific T lymphocytes has long been proposed and performed for treatment of cancers refractory to conventional therapeutic approaches (CitationYee 2013). The common modalities for adoptive transfer of autologous or allogeneic T cells encounter some problems, including the time consuming processes for development of desired cell populations, short in vivo half-life, and self-MHC restriction of T cells for activation (CitationRiether et al. 2013). Advances in genetic engineering of T cells has given rise to introduce of chimeric antigen receptor (CAR) T cells or “T-body” technology (CitationDotti et al. 2009). A CAR consists of an extracellular recognition domain, including variable domains of heavy (VH) and light (VL) chains derived from tumor antigen-specific B cells. The recognition unit extends to a spacer and then the transmembrane domain and intracellular signaling motifs derived from stimulatory molecules, for example, CD28, CD3 ζ, 4-1BB, and OX40 (CitationCartellieri et al. 2010, CitationHan et al. 2013). According to cytoplasmic signaling domains built into a CAR, there are three generations of CARs containing intracellular motifs of one, two, or three of the above-mentioned molecules (). Genes coding for VH and VL domains and also signaling motifs (collectively, scFV) are cloned into a retroviral vector and transfected in T lymphocytes derived from the patient's body. Because of the nature of retroviral vectors, inserted genes are integrated into the genome of T cells and these cells bear their own “artificial” specificity throughout their lifecycle. After re-transfusion of engineered T cells, they recognize tumor-specific antigens (TSAs) and respond with activation, proliferation, and exertion of effector functions to kill tumor cells. Antigen recognition by CARs brings along some advantages including non-MHC restricted recognition and T cell proliferation, the recognition of not only protein but other types of antigens, capability of giving desired specificity to both CD4+ and CD8+ T cells, and lower probability of eliciting unwanted responses especially autoimmunity (CitationSadelain et al. 2013).
Figure 2. Different generations of CARs used in therapeutic approaches.
Several antigenic targets of hematologic and solid tumors have been selected, and CAR T cells have been designed and used in murine models and also in clinical trials (CitationKershaw et al. 2013). The most frequent target for CAR T cell therapy has been CD19 in several CD19-expressing malignancies. CTL019, formerly called CART19, is one of the most promising CAR T cell therapies designed for treatment of ALL. CT019 resulted in complete remission in a study of two children with relapsed chemotherapy- refractory ALL (CitationDavila et al. 2013). There are also ongoing studies on application of CTL019 therapy in B-CLL.
Cancer vaccines
History
Vaccination is a very old medical procedure that induces the immune system's functions and brings a long-lasting immunologic memory to protect the body against foreign elements such as microbes (CitationLambert et al. 2005). Curiosity in vaccination against malignancy commenced around the 1900s, when the effectiveness of microbial vaccines had been already proved. The thought was rational: to apply killed or inactive malignant cells using a similar approach, but in the context of a tumor. Recently, advances in active immunotherapy for prevention and treatment of cancers has been emphasized (CitationJager et al. 2002). By priming or boosting the immune system's natural capability, cancer vaccines are effective medicines that are categorized in a class of therapeutics known as biologic response modifiers (BRM). Cancer vaccines achieve this effect by introducing single or combined molecules known as tumor antigens into the immune system. Dendritic cells, the most proficient APCs, can take-up tumor antigens, and depending on the environmental and inflammatory conditions, present the antigens at the tumor sites or at lymphoid organs to prime, sustain, or abrogate the tumor-specific immune response. Due to the self-origin of tumor cells and the natural immuno-evasive and suppressive mechanisms of tumor cells, a few cancer vaccines have been approved for clinical use (CitationTabi and Man 2006).
There are two classes of tumor antigens, including abnormal self-antigens (ASAs) and TSAs (CitationNeville et al. 1975). ASAs are usually embryonal and developmental antigens not normally expressed in adult tissues, normal proteins with unusual glycosylation patterns, or overexpressed self-antigens. TSAs spawn following somatic mutations or damages in the germline DNA that lead to errors in the mature mRNA or to fusion proteins (CitationFinn 2006). Not all such mutations change the immunogenicity of tumor cells, and the question of the degree to which TSAs comply with the requirements for fitting to the MHC antigen-binding site (CitationSegal et al. 2008) remains unanswered. Recently, enormous TSAs have been discovered which may be fitting for antigen-defined cancer vaccines and include antigens discovered as deriving from somatic mutations in oncogenes or tumor suppressor genes (for example, RAS, BCR/ABL, BRCA1,2, HER2,3 and P53), cancer/testis antigen (NY-ESO-1), developmental antigens (for example, MAGE, tyrosinase, gp100), and overexpressed antigens (for example, oncofetal antigens, carcinoembryonic antigen, α-fetoprotein) (CitationGreiner et al. 2002, CitationDe Giovanni et al. 2004, CitationSobol 2006, CitationTheobald et al. 1995).
Great numbers of potential tumor antigens have been described by the Academy of Cancer Immunology (http://www.cancerimmunity.org/peptidedatabase/Tcellepitopes.htm). Moreover, the Cancer Genome Atlas (TCGA) has recently revealed large numbers of potential tumor antigens in different types of solid tumors.
Despite defined tumor antigen-based vaccines (for example, DNA and peptide-based vaccines), some more recent and unconventional vaccine platforms (for example, tumor-cell-based vaccines, allogeneic tumor heat-shock proteins) depend on the construction of shared antigens between similar tumor cells and therefore do not require identification of certain tumor antigens in advance (CitationSchreiber et al. 2010).
Cancer-preventive and treatment vaccines
There are two basic classes of cancer vaccines. Preventive vaccines, which are supposed to prevent initiation of malignant transformation in normal cells of healthy subjects, and treatment vaccines, which are intended to treat an established cancer by intensification of the immune system's antitumor capability (CitationLollini et al. 2006). Three cancer-preventive vaccines have been released in the United States, and one cancer treatment vaccine also has recently become available. Cancer-preventive vaccines target infectious agents that cause or are associated with the development of cancer (CitationDoorbar 2006, CitationFrazer et al. 2007). The FDA has just approved Gardasil, manufactured by Merck & Company, and Cervarix, manufactured by GlaxoSmithKline, that protect against infection by HPV—types 16 and 18—that cause approximately 70 percent of all cases of cervical cancer (CitationDoorbar 2006). The FDA has also approved a cancer-preventive vaccine that immunizes against the HBV infection which can lead to hepatocellular carcinoma. The original HBV vaccine was approved in early 1980s, making it the first-in-class cancer preventive vaccine to be successfully developed. Today, most newborns in the United States are vaccinated against HBV (CitationYaddanapudi et al. 2013).
Cancer treatment vaccines are supposed to treat tumors that have already established and are well-grown. They have been proven more difficult and challenging to produce than cancer preventive vaccines (CitationRosenberg et al. 2004). Cancer treatment vaccines must meet more demands. Actually, they must stimulate immune responses that are specific and strong enough against the proper target antigen and cells, to defeat the barriers of immuno-evasion and suppression that cancer cells exploit to protect themselves from immune responses. Using whole tumor cell extract, tumor antigens, whole tumor cells, as well as costimulatory molecules and TSA-encoding recombinant DNA, cancer treatment vaccines are poised to delay or hold back transformed cell growth and to block tumor recurrence. Scientists are developing methods of immunotherapy that can be used to prime/boost tumor-specific immune responses that are currently being evaluated as treatment vaccines (CitationAntonia et al. 2004, CitationYaddanapudi et al. 2013).
In 2010, the FDA approved the first-in-class cancer treatment vaccine, sipuleucel-T (Provenge®, manufactured by Dendreon), for treatment of hormone-refractory prostate cancer and metastatic prostate cancer (CitationAntonia et al. 2004). It is designed to trigger an immune response to prostatic acid phosphatase (PAP), also known as kallikrein-3 (KLK3), an over-expressed tumor antigen (CitationKantoff et al. 2010). Provenge is generated by isolating APCs and cultured with a PAP linked to GM-CSF. The approval of Provenge has inspired more efforts toward the development of similar therapies. However, what has become apparent is that the most effective form of vaccines against cancer would also act as preventive agents; although rather than protecting against malignancy, vaccines would be used to prevent relapse in minimal disease settings. The reason that this clinical setting seems better than the bulky cancer setting is that tumors alter many immunological components and environments, which directly hinders immune response to the vaccine, increases the tumor burden, and leads to lowered effectiveness of the vaccine (CitationNestle et al. 2005).
Strategies for Cancer vaccination
Identifying the right antigen, the right adjuvant, and the right immune responses are the common challenges facing cancer vaccination. Several types of vaccines have been proved to generate tumor-antigen-specific immunity including peptide, protein, whole tumor cells, DC, DNA, and viral, each with its own potencies and limitations (CitationNestle et al. 2005, CitationPijpers et al. 2005).
Peptide-based vaccines
A peptide cancer vaccine is kind of subunit vaccine in which a specific peptide of the complex tumor antigen is used for immunization. Tumor-specific peptides are identified based on binding capacity to specific molecules of HLA-I or II (CitationPijpers et al. 2005). The advantage of targeting the HLA class I, but not class II molecules, is that a high percentage (up to 35–50%) of the population may carry a copy of individual HLA-I (CitationYaddanapudi et al. 2013). The peptides that bind to HLA class I enclose unique sequences, mounting the numbers of sequence that would need to be built into the vaccine with adequate population coverage. Studies have just shown that an individual HLA-DR peptide may bind to numerous HLA-DR subtypes (CitationDisis et al. 2002). Therefore, it might take fewer HLA class II peptides, compared to peptides of HLA class I, to prepare a wide range vaccines. Despite protein vaccines, the greater cost benefits, ease of synthesis, formulation, and delivery are further advantages of peptide-based cancer vaccines. One probable difficulty with using specific peptide cancer vaccines is the occurrences of antigen-loss variants of tumor cells (CitationZhou et al. 2004, CitationSanchez-Perez et al. 2005). Polyvalent tumor antigens such as allogeneic whole tumor cell vaccines, which may minimize the risk of antigen loss variants and increase population coverage, seem to offer advantages when applied. (CitationYajima et al. 2005).
Whole tumor cell vaccines
Whole tumor cell vaccination is an effective procedure of injecting attenuated or killed tumor cells, usually along with costimulatory compounds such as cytokines (CitationParmiani et al. 2011, CitationLi et al. 2007). The procedure falls under the three basic classes including autologous, allogeneic and gene-modified vaccines. In contrast to specific peptide vaccines, whole tumor cell vaccination allows for the immune system's natural ability to recognize most immunogenic TSAs, which obviates the need for MHC restriction-specific epitope identification. However, possible breakage in self-tolerance to normal molecules in the presence of costimulatory adjuvants might result in autoimmune responses. In a gene-modified approach, modification of inactive melanoma cells to secrete immunostimulatory molecules, such as GM-CSF, was shown to improve tumor antigen presentation through DC and macrophage-evident recruitment, production of tumor-specific CD4+ and CD8+ T cells, NKT-cells and antibodies for successful tumor rejection (CitationDranoff 2003). Autologous whole tumor cell approaches are currently being evaluated to treat acute myeloid leukemia and metastatic non-small cell lung carcinoma (CitationCheuk et al. 2006, CitationSalgia et al. 2003).
Heat-shock protein vaccines
Heat-shock proteins (HSPs) carrying multiple undefined immunogenic tumor antigens can be purified from a patient's tumor cells and used as a polyvalent autologous cancer vaccine preparation (CitationSrivastava 2005). The significance of HSPs in tumor antigen presentation and cross-presentation still remains undiscovered. Some reports agree on the opinion that HSPs can bind and present tumor antigens to APCs through HLA-I and II molecules, to activate tumor-specific CD8+ and CD4+ T cells (CitationSrivastava 2005). The effectiveness of HSP vaccines lies in the ability of HSPs to concurrently serve as adjuvants and stimulate both innate and adaptive immune responses. Upon engagement of surface HSP receptors such as c-type lectin receptors and scavenger receptors, DC undergo a maturation process that enables them to become potent APCs (CitationBinder et al. 2000). Potent immune responses and evident lesion regression was reported in a late phase II clinical trials using the Hsp7-HPV 16 fusion protein vaccine in women with advanced cervical cancer (CitationRoman et al. 2007).
Anti-idiotypic antibody-based vaccines
Injection of tumor-specific mAbs may result in the formation of autologous antibodies against the original vaccine (CitationMocellin et al. 2004). The latter antibodies, known as anti-idiotypic antibodies, are specific for idiotopes of the original mAb. These antibodies, in an adjuvant combination, are used as a cancer vaccine (CitationRico and Hall 1989). An example of anti-idiotype cancer vaccine is Racotumomab. Racotumomab induces and targets immune response to a type of ganglioside, N-glycolil (NGc) GM3 (NGcGM3), expressed on the surface of lung, breast, melanoma tumor cells (CitationVazquez et al. 2012). An additional well-studied tumor antigen is the tumor-specific idiotype expressed on B cell lymphomas (CitationRico and Hall 1989). The vaccines studied are comprised of tumor-derived Ig containing tumor-specific idiotypes. The significance of anti-idiotypic vaccines is that it allows effective immunization against non-protein antigens, such as tumor-specific carbohydrate or lipid antigens, to draw an effective T cell memory.
Dendritic cell-based vaccines
DC therapy or DC vaccine is a recent, safe, and promising class of cancer treatment and prevention therapy even in advanced cancer patients who have failed all the possible therapies (CitationPalucka and Banchereau 2013). As the most competent activators of naive T cells, taking DCs in cancer drugs to induce the effective tumor antigen-specific response makes great sense for tumor biologists (CitationPalucka and Banchereau 2013, CitationSteinman and Banchereau 2007). Several DC-based cancer vaccines have been developed to date, including DC loaded with tumor peptides or whole proteins (CitationLi et al. 2000, CitationTimmerman et al. 2002), DC pulsed with tumor antigen RNA or DNA (CitationBoczkowski et al. 2000, CitationMilazzo et al. 2003, CitationNencioni et al. 2003), DC transduced with viral vectors such as lentiviruses, retroviruses, fowlpox, adenoviruses and alphaviruses (CitationKim et al. 1998, CitationCaley et al. 1997) whole-killed and protein extract of tumor cells (CitationGalea-Lauri et al. 2004, CitationChen et al. 2001, CitationFerlazzo et al. 2000), and DC-fused with tumor cells using hybrid technology (CitationGong et al. 2000, CitationChen et al. 2000, CitationGarcia-Marquez et al. 2013). Although DC-based vaccines have shown promising results in several preclinical and clinical settings, choosing the proper DC functional sub-population with different functions and capacities is a great matter to deal with. Each functional subpopulation of DC has a unique capability of activating or suppressing different functional T CD4 + subsets (CitationFeili-Hariri et al. 2005, CitationPalucka and Banchereau 2013, CitationStrioga et al. 2013).
DNA and viral vector-based vaccines
DNA vaccines are designed to produce an immunological response against certain antigens by injecting the genetically engineered DNA encoding corresponding antigens. DNA vaccination has unique advantages over other vaccination procedures, including the capability to induce a broad spectrum of immune responses, benefits afforded by their ease of production and low cost, and the fact that the information pertaining to the HLA-I and II genotypes (CitationStevenson et al. 2004, CitationStevenson 2004) is not required. Similar to protein-based vaccines, DNA vaccines depend on antigen processing and presentation by APCs (CitationDonnelly et al. 2005, CitationZhou et al. 2004). DNA vaccines are supposed to deliver a gene of particular tumor antigens to the body as a bacterial vector. The first demonstration of a plasmid-induced immune response was observed when mice inoculated with a plasmid- expressing human growth hormone elicited antibodies instead of altering growth (CitationTang et al. 1992). In practice, the infected host cells express the particular tumor antigen, drain into the lymph node, and eventually, after being taken up by DCs and presented to T cells or being recognized by B cells, trigger a broad range of immune responses (CitationYu and Finn 2006). In contrast to peptide-based vaccines that draw on a specific arm of immunity, DNA vaccines are characterized by activation of a broad spectrum of effector arms of the immune system (CitationZhou et al. 2005). The risk of integration with the host genome and thereby affecting genes controlling growth and survival, the possibility of antibody production against DNA, and the limitation to protein tumor antigen are downsides of DNA vaccines (CitationRobinson and Pertmer 2000). However, application of RNA instead can obviate the possibility of integration (CitationHess et al. 2006, CitationHeiser et al. 2000). DNA vaccine platforms can be made more immunogenic by inserting and encoding DNA into viral vectors. Live recombinant viral vaccines have been studied as cancer vaccines for years (CitationHarrop and Carroll 2006). By mimicking a natural infection and offering the danger signals required for full activation of DC, viral vectors present a promising strategy for tumor antigen delivery (CitationDraper and Heeney 2010). The first trial with viral vectors was vaccinia, over two decades ago (CitationMackett et al. 1982), and numerous viral vectors have been constructed on the poxviruses, such as avian poxviruses, fowlpox, modified vaccinia virus Ankara (MVA), canarypox, recombinant adenoviruses, and herpes virus (Citationde Bruyn et al. 2004, CitationTriozzi et al. 2005, CitationRosenberg et al. 1998).
Particle-based vaccine
Microparticles, emulsions, immune-stimulating complexes (ISCOM), liposomes, virosomes, and virus-like-particles (VLPs), are particulate vehicles for antigen that are increasingly being applied in vaccine formulations as carriers that deliver the tumor antigen to DC, in a much more immunogenic form. Particle platforms provide several advantages, such as sharing a similarity in size with the pathogens, and the capacity to carry multiple copies of the tumor antigen on the surface of the particle, which can be as effective as those internalized by DC, macrophages, and B cells (CitationNewman et al. 2002, CitationRandolph et al. 1999). Moreover, danger signals, and costimulators such as HSP and unmethylated CpG DNA, could be incorporated with the tumor antigens during formulation to boost the activation of DC and elicit optimal adaptive immune response (CitationO’Hagan et al. 2006).
Combinational therapy
Although recent clinical approvals on protective and therapeutic cancer vaccines represent major milestones, the optimal achievement of cancer therapy will likely benefit from a combination of cancer vaccines with other immunotherapeutic and non-immunotherapeutic approaches such as mAbs, chemotherapy, radiation therapy, and surgery. It is assumed that following a cancer vaccine (‘prime’) with other immunotherapeutics and/or non-immunotherapeutics (‘boost’) may provide the best-in-class cancer therapy regimen (CitationFinn 2008). Several preclinical and clinical settings for combinational therapy have emerged. Cytotoxicity of HER-2 specific T cells is increased against tumor cells pretreated with trastuzumab (Citationzum Buschenfelde et al. 2002). It is assumed that the antibody causes the internalization and degradation of HER-2, resulting in increased presentation of HER-2-MHC and eventually greater activation and expansion of HER-2-specific T cells. Clinical trials are currently underway to test for potentially improved efficacy using this combination. Using agents and protocols to deplete or inhibit circulating Treg such as cyclophosphamide, anti-CTLA4, and CD25-targeted agents such as Denileukin diftitox-enhanced immune-based therapies (CitationPhan et al. 2003, CitationZou 2005).
Targeting cancer stem cell
Accumulating evidence suggests that a biologically unique subpopulation of tumor cells with undifferentiated and stem cell-like properties lies behind the formation and progression of tumors (CitationSchächinger et al. 2004, CitationWollert et al. 2004). These infrequent cells can be distinguished from the vast majority of tumor bulk cells by their exclusive ability to initiate and perpetuate the growth of a malignant cell population indefinitely (CitationNguyen et al. 2012, CitationSchulenburg et al. 2010). They are widely named “cancer stem cells” or tumor-initiating cells (TICs). Actually, these biologically unique subsets of cells (CSCs) are named and defined by their demonstrated ability to regenerate, progressively growing the tumor. The growth consists of cells resembling those in the original tumor (self-replicating ability) (CitationSarry et al. 2011), as studied through serial transplantations in immunodeficient non-obese diabetic (NOD)/severe combined immunodeficient (SCID) mice.
The CSC concept places considerable significance on clinical cancer therapy, as it throws light on that current therapeutic strategies that are generally developed to target the cancer bulk rather the CSCs as the seed of tumors. Moreover it may explain the reason why many treatments seem to be effective primarily but fail later due to ineffectiveness of current therapies on the rarer and grossly invisible populations of CSCs, which remain safe and sound to re-initiate tumor formation (CitationBao et al. 2006, CitationChen et al. 2007).
On the whole, this study emphasizes that the ultimate goal in the treatment of cancer is elimination of CSCs. To date, most efforts have focused on targets specifically expressed in CSCs, to develop new strategies for CSC elimination. Targeting surface molecules was the first that struck us as being a sensible strategy to distinguish CSCs from bulk tumor cells. However, surface molecules are expressed on the normal stem cells of the tissue as well, heightening the necessity of identification of more specific molecules that are exclusively expressed on CSCs (CitationJordan et al. 2006, CitationEssers and Trumpp 2010).
CD133+ cancer cells of the brain, colon, lung, and pancreas, CD44+CD24low or CD44+CD24− human breast cancer cells, CD47+ bladder, CD90+ liver, ABCB5+ (ATP-binding cassette sub-family B member 5) of melanoma and CD34+CD38low or CD34+CD38−of many human AML cancer cells are well characterized and accepted as CSCs (CitationTaussig et al. 2008, CitationNguyen et al. 2012, CitationRiether et al. 2013).
Immunotherapy is now considered to be a reasonable strategy to directly attack CSCs and eradicate quiescent CSCs. Candidates for immunotherapy of CSCs include CSCs mAb therapy, activated cytotoxic CD8 + T cells (CTLs), and NK cells specific for CSCs; another possibility is to force CSCs into the cell-cycle by breaking their dormancy, followed by conventional cytotoxic chemotherapy () (CitationRiether et al. 2013).
Table II. Targeting cancer stem cells (CSCs) using immunotherapy.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abrams DI, Volberding PA. 1986. Alpha interferon therapy of AIDS-associated Kaposi's sarcoma. Semin Oncol. 13:43–47.
- Ahmed S, Rai KR. 2003. Interferon in the treatment of hairy-cell leukemia. Best Pract Res Clin Haematol. 16:69–81.
- Antonia S, Mule JJ, Weber JS. 2004. Current developments of immunotherapy in the clinic. Curr Opin Immunol. 16:130–136.
- Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. 1999. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 17:2105–2116.
- Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. 2010. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self Nonself. 1:314–322.
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. 2006. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 444:756–760.
- Bhutia SK, Mallick SK, Maiti TK. 2010. Tumour escape mechanisms and their therapeutic implications in combination tumour therapy. Cell Biol Int. 34:553–563.
- Binder RJ, Han DK, Srivastava PK. 2000. CD91: a receptor for heat shock protein gp96. Nat Immunol. 1:151–155.
- Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. 2000. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res. 60:1028–1034.
- Bonifazi F, De Vivo A, Rosti G, Guilhot F, Guilhot J, Trabacchi E, et al. 2001. Chronic myeloid leukemia and interferon-alpha: a study of complete cytogenetic responders. Blood. 98:3074–3081.
- Borrebaeck CA. 1989. Strategy for the production of human monoclonal antibodies using in vitro activated B cells. J Immunol Methods. 123:157–165.
- Breccia M, Lo-Coco F. 2011. Gemtuzumab ozogamicin for the treatment of acute promyelocytic leukemia: mechanisms of action and resistance, safety and efficacy. Expert Opin Biol Ther. 11: 225–234.
- Burnet M. 1957. Cancer—A Biological Approach: III. Viruses associated with neoplastic conditions. IV. Practical applications. Br Med J. 1:841.
- Caley IJ, Betts MR, Irlbeck DM, Davis NL, Swanstrom R, Frelinger JA, Johnston RE. 1997. Humoral, mucosal, and cellular immunity in response to a human immunodeficiency virus type 1 immunogen expressed by a Venezuelan equine encephalitis virus vaccine vector. J Virol. 71:3031–3038.
- Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, et al. 2010. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol. 2010: 956304.
- Chen D, Koido S, Li Y, Gendler S, Gong J. 2000. T cell suppression as a mechanism for tolerance to MUC1 antigen in MUC1 transgenic mice. Breast Cancer Res Treat. 60:107–115.
- Chen MS, Woodward WA, Behbod F, Peddibhotla S, Alfaro MP, Buchholz TA, Rosen JM. 2007. Wnt/β-catenin mediates radiation resistance of Sca1 + progenitors in an immortalized mammary gland cell line. J Cell Sci. 120:468–477.
- Chen Z, Moyana T, Saxena A, Warrington R, Jia Z, Xiang J. 2001. Efficient antitumor immunity derived from maturation of dendritic cells that had phagocytosed apoptotic/necrotic tumor cells. Int J Cancer. 93:539–548.
- Cheuk AT, Chan L, Czepulkowski B, Berger SA, Yagita H, Okumura K, et al. 2006. Development of a whole cell vaccine for acute myeloid leukaemia. Cancer Immunol Immunother. 55:68–75.
- Costello RT, Gastaut JA, Olive D. 1999. Tumor escape from immune surveillance. Arch Immunol Ther Exp (Warsz). 47:83–88.
- Davila ML, Kloss CC, Gunset G, Sadelain M. 2013. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One. 8:e61338.
- de Bruyn G, Rossini AJ, Chiu YL, Holman D, Elizaga ML, Frey SE, et al. 2004. Safety profile of recombinant canarypox HIV vaccines. Vaccine. 22:704–713.
- De Giovanni C, Nicoletti G, Landuzzi L, Astolfi A, Croci S, Comes A, et al. 2004. Immunoprevention of HER-2/neu transgenic mammary carcinoma through an interleukin 12-engineered allogeneic cell vaccine. Cancer Res. 64:4001–4009.
- Disis ML, Gooley TA, Rinn K, Davis D, Piepkorn M, Cheever MA, et al. 2002. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol. 20:2624–2632.
- Donnelly JJ, Wahren B, Liu MA. 2005. DNA vaccines: progress and challenges. J Immunol. 175:633–639.
- Doorbar J. 2006. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond). 110:525–541.
- Dotti G, Savoldo B, Brenner M. 2009. Fifteen years of gene therapy based on chimeric antigen receptors: “are we nearly there yet?”. Hum Gene Ther. 20:1229–1239.
- Dranoff G. 2003. GM-CSF-secreting melanoma vaccines. Oncogene. 22:3188–3192.
- Draper SJ, Heeney JL. 2010. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol. 8:62–73.
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. 2002. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 3:991–998.
- Dutcher JP. 2011. High-dose interleukin-2 therapy for metastatic renal cell carcinoma and metastatic melanoma: still the standard. Oncology (Williston Park). 25:427–428.
- Ehrlich PR, Himmelweit F. 1956. The collected papers of Paul Ehrlich. London:Pergamon Press.
- Essers MAG, Trumpp A. 2010. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 4:443–450.
- Feili-Hariri M, Falkner DH, Morel PA. 2005. Polarization of naive T cells into Th1 or Th2 by distinct cytokine-driven murine dendritic cell populations: implications for immunotherapy. J Leukoc Biol. 78:656–664.
- Ferlazzo G, Semino C, Spaggiari GM, Meta M, Mingari MC, Melioli G. 2000. Dendritic cells efficiently cross-prime HLA class I-restricted cytolytic T lymphocytes when pulsed with both apoptotic and necrotic cells but not with soluble cell-derived lysates. Int Immunol. 12:1741–1747.
- Finn OJ. 2006. Human tumor antigens, immunosurveillance, and cancer vaccines. Immunol Res. 36:73–82.
- Finn OJ. 2008. Cancer immunology. N Engl J Med. 358:2704–2715.
- Finn OJ. 2012. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 23 Suppl 8:viii6–9.
- Flynn S, Stockinger B. 2003. Tumor and CD4 T-cell interactions: tumor escape as result of reciprocal inactivation. Blood. 101:4472–4478.
- Frazer IH, Lowy DR, Schiller JT. 2007. Prevention of cancer through immunization: Prospects and challenges for the 21st century. Eur J Immunol. 37 Suppl 1:S148–155.
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. 1996. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 2:1096–1103.
- Galea-Lauri J, Wells JW, Darling D, Harrison P, Farzaneh F. 2004. Strategies for antigen choice and priming of dendritic cells influence the polarization and efficacy of antitumor T-cell responses in dendritic cell-based cancer vaccination. Cancer Immunol Immunother. 53:963–977.
- Garcia-Marquez MA, Shimabukuro-Vornhagen A, Theurich S, von Bergwelt-Baildon M. 2013. Vaccination with dendritic cell-tumor fusion cells in multiple myeloma patients: a promising strategy? Immunotherapy. 5:1039–1042.
- Gong J, Nikrui N, Chen D, Koido S, Wu Z, Tanaka Y, et al. 2000. Fusions of human ovarian carcinoma cells with autologous or allogeneic dendritic cells induce antitumor immunity. J Immunol. 165:1705–1711.
- Greiner JW, Zeytin H, Anver MR, Schlom J. 2002. Vaccine-based therapy directed against carcinoembryonic antigen demonstrates antitumor activity on spontaneous intestinal tumors in the absence of autoimmunity. Cancer Res. 62:6944–6951.
- Group OMTS. 1985. A randomized clinical trial of OKT3 monoclonal antibody for acute rejection of cadaveric renal transplants. N Engl J Med. 313:337–342.
- Han EQ, Li XL, Wang CR, Li TF, Han SY. 2013. Chimeric antigen receptor-engineered T cells for cancer immunotherapy: progress and challenges. J Hematol Oncol. 6:47.
- Harding FA, Stickler MM, Razo J, DuBridge RB. 2010. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2:256–265.
- Harrop R, Carroll MW. 2006. Viral vectors for cancer immunotherapy. Front Biosci. 11:804–817.
- Hauschild A. 2009. Adjuvant interferon alfa for melanoma: new evidence-based treatment recommendations? Curr Oncol. 16:3–6.
- Heiser A, Dahm P, Yancey DR, Maurice MA, Boczkowski D, Nair SK, et al. 2000. Human dendritic cells transfected with RNA encoding prostate-specific antigen stimulate prostate-specific CTL responses in vitro. J Immunol. 164:5508–5514.
- Hess PR, Boczkowski D, Nair SK, Snyder D, Gilboa E. 2006. Vaccination with mRNAs encoding tumor-associated antigens and granulocyte-macrophage colony-stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol Immunother. 55:672–683.
- Huang SM, Bock JM, Harari PM. 1999. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 59:1935–1940.
- Igney FH, Krammer PH. 2002. Immune escape of tumors: apoptosis resistance and tumor counterattack. J Leukoc Biol. 71:907–920.
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. 2002. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 99:12293–12297.
- Jacobs SA. 2007. Yttrium ibritumomab tiuxetan in the treatment of non-Hodgkin's lymphoma: current status and future prospects. Biologics. 1:215–227.
- Jager E, Jager D, Knuth A. 2002. Clinical cancer vaccine trials. Curr Opin Immunol. 14:178–182.
- Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. 1986. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 321:522–525.
- Jordan CT, Guzman ML, Noble M. 2006. Cancer stem cells. N Engl J Med. 355:1253–1261.
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. 2010. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 363:411–422.
- Kershaw MH, Westwood JA, Darcy PK. 2013. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 13:525–541.
- Kim CJ, Cormier J, Roden M, Gritz L, Mazzara GP, Fetsch P, et al. 1998. Use of recombinant poxviruses to stimulate anti-melanoma T cell reactivity. Ann Surg Oncol. 5:64–76.
- Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. 2005. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 11:1010–1020.
- Kim KJ, Li B, Houck K, Winer J, Ferrara N. 1992. The vascular endothelial growth factor proteins: identification of biologically relevant regions by neutralizing monoclonal antibodies. Growth Factors. 7:53–64.
- Kim R, Emi M, Tanabe K. 2007. Cancer immunoediting from immune surveillance to immune escape. Immunology. 121:1–14.
- Kirkwood J. 2002. Cancer immunotherapy: the interferon-α experience. Semin Oncol. 29:18–26.
- Kohler G, Milstein C. 1975. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 256:495–497.
- Kyi C, Postow MA. 2014. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett. 588:368–376.
- Lambert PH, Liu M, Siegrist CA. 2005. Can successful vaccines teach us how to induce efficient protective immune responses? Nat Med. 11:S54–62.
- Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C, Shepard HM. 1993. Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer Immunol Immunother. 37:255–263.
- Li H, Jiang HJ, Ma MQ, Wei F, An XM, Ren XB. 2007. Vaccination with allogeneic GM-CSF gene-modified lung cancer cells: antitumor activity comparing with that induced by autologous vaccine. Cancer Biother Radiopharm. 22:790–798.
- Li Y, Bendandi M, Deng Y, Dunbar C, Munshi N, Jagannath S, et al. 2000. Tumor-specific recognition of human myeloma cells by idiotype-induced CD8(+) T cells. Blood. 96:2828–2833.
- Lollini PL, Cavallo F, Nanni P, Forni G. 2006. Vaccines for tumour prevention. Nat Rev Cancer. 6:204–216.
- Lonberg N. 2008. Fully human antibodies from transgenic mouse and phage display platforms. Curr Opin Immunol. 20:450–459.
- Mackett M, Smith GL, Moss B. 1982. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc Natl Acad Sci U S A. 79:7415–7419.
- Maeurer MJ, Gollin SM, Martin D, Swaney W, Bryant J, Castelli C, et al. 1996. Tumor escape from immune recognition: lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. J Clin Invest. 98:1633–1641.
- Mellor AL, Munn D, Chandler P, Keskin D, Johnson T, Marshall B, et al. 2003. Tryptophan catabolism and T cell responses. Adv Exp Med Biol. 527:27–35.
- Mesa C, Fernandez LE. 2004. Challenges facing adjuvants for cancer immunotherapy. Immunol Cell Biol. 82:644–650.
- Meyer JP, Persad R, Gillatt DA. 2002. Use of bacille Calmette-Guerin in superficial bladder cancer. Postgrad Med J. 78:449–454.
- Meyer T, Surber C, French LE, Stockfleth E. 2013. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin Investig Drugs. 22:149–159.
- Milazzo C, Reichardt VL, Muller MR, Grunebach F, Brossart P. 2003. Induction of myeloma-specific cytotoxic T cells using dendritic cells transfected with tumor-derived RNA. Blood. 101:977–982.
- Minasian LM, Motzer RJ, Gluck L, Mazumdar M, Vlamis V, Krown SE. 1993. Interferon alfa-2a in advanced renal cell carcinoma: treatment results and survival in 159 patients with long-term follow-up. J Clin Oncol. 11:1368–1375.
- Mittler RS, Foell J, Mccausland M, Strahotin S, Niu L, Bapat A, Hewes LB. 2004. Anti-CD137 antibodies in the treatment of autoimmune disease and cancer. Immunol Res. 29:197–208.
- Mocellin S, Mandruzzato S, Bronte V, Lise M, Nitti D. 2004. Part I: Vaccines for solid tumours. Lancet Oncol. 5:681–689.
- Mocellin S, Marincola FM, Young HA. 2005. Interleukin-10 and the immune response against cancer: a counterpoint. J Leukoc Biol. 78:1043–1051.
- Moran AE, Kovacsovics-Bankowski M, Weinberg AD. 2013. The TNFRs OX40, 4–1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol. 25:230–237.
- Nencioni A, Muller MR, Grunebach F, Garuti A, Mingari MC, Patrone F, et al. 2003. Dendritic cells transfected with tumor RNA for the induction of antitumor CTL in colorectal cancer. Cancer Gene Ther. 10:209–214.
- Nestle FO, Tonel G, Farkas A. 2005. Cancer vaccines: the next generation of tools to monitor the anticancer immune response. PLoS Med. 2:e339.
- Neuberger MS, Williams GT, Mitchell EB, Jouhal SS, Flanagan JG, Rabbitts TH. 1985. A hapten-specific chimaeric IgE antibody with human physiological effector function. Nature. 314:268–270.
- Neville AM, Mackay AM, Westwood J, Turberville C, Laurence DJ. 1975. Human tumour-associated and tumour-specific antigens: some concepts in relation to clinical oncology. J Clin Pathol Suppl (Assoc Clin Pathol). 6:102–112.
- Newman KD, Elamanchili P, Kwon GS, Samuel J. 2002. Uptake of poly(D,L-lactic-co-glycolic acid) microspheres by antigen-presenting cells in vivo. J Biomed Mater Res. 60:480–486.
- Newstead C. 1998. Assessment of risk of cancer after renal transplantation. Lancet. 351:610–611.
- Nguyen LV, Vanner R, Dirks P, Eaves CJ. 2012. Cancer stem cells: an evolving concept. Nat Rev Cancer. 12:133–143.
- O’Hagan DT, Singh M, Ulmer JB. 2006. Microparticle-based technologies for vaccines. Methods. 40:10–19.
- Oldfield V, Keating GM, Perry CM. 2005. Imiquimod: in superficial basal cell carcinoma. Am J Clin Dermatol. 6:195–200; discussion 201–202.
- Oldham RK. 1983. Monoclonal antibodies in cancer therapy. J Clin Oncol. 1:582–590.
- Pageau SC. 2009. Denosumab. MAbs. 1:210–215.
- Palucka K, Banchereau J. 2013. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 39:38–48.
- Pardoll DM. 2012. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
- Parmiani G, Pilla L, Maccalli C, Russo V. 2011. Autologous versus allogeneic cell-based vaccines? Cancer J. 17:331–336.
- Pasche B. 2001. Role of transforming growth factor beta in cancer. J Cell Physiol. 186:153–168.
- Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. 2003. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 100:8372–8377.
- Pijpers F, Faint R, Saini N. 2005. Therapeutic cancer vaccines. Nat Rev Drug Discov. 4:623–624.
- Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. 1999. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 11:753–761.
- Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, et al. 1994. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 83:435–445.
- Rico MJ, Hall RP 3rd. 1989. Anti-idiotypic antibodies as vaccine candidates. The immune network. Arch Dermatol. 125:271–275.
- Riether C, Schurch C, Ochsenbein AF. 2012. Modulating CD27 signaling to treat cancer. Oncoimmunology. 1:1604–1606.
- Riether C, Schurch C, Ochsenbein AF. 2013. From “magic bullets” to specific cancer immunotherapy. Swiss Med Wkly. 143:w13734.
- Robinson HL, Pertmer TM. 2000. DNA vaccines for viral infections: basic studies and applications. Adv Virus Res. 55:1–74.
- Roman LD, Wilczynski S, Muderspach LI, Burnett AF, O’Meara A, Brinkman JA, et al. 2007. A phase II study of Hsp-7 (SGN-00101) in women with high-grade cervical intraepithelial neoplasia. Gynecol Oncol. 106:558–566.
- Rosenberg SA, Yang JC, Restifo NP. 2004. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 10:909–915.
- Rosenberg SA, Zhai Y, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, et al. 1998. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J Natl Cancer Inst. 90:1894–1900.
- Rutar FJ, Augustine SC, Kaminski MS, Wahl RL, Siegel JA, Colcher D. 2001. Feasibility and safety of outpatient Bexxar therapy (tositumomab and iodine I 131 tositumomab) for non-Hodgkin's lymphoma based on radiation doses to family members. Clin Lymphoma. 2:164–172.
- Sadelain M, Brentjens R, Riviere I. 2013. The basic principles of chimeric antigen receptor design. Cancer Discov. 3:388–398.
- Salazar-Onfray F. 1999. Interleukin-10: a cytokine used by tumors to escape immunosurveillance. Med Oncol. 16:86–94.
- Salgia R, Lynch T, Skarin A, Lucca J, Lynch C, Jung K, et al. 2003. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J Clin Oncol. 21:624–630.
- Sanchez-Perez L, Kottke T, Diaz RM, Ahmed A, Thompson J, Chong H, et al. 2005. Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo. Cancer Res. 65:2009–2017.
- Sarry J-E, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. 2011. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 121:384.
- Schächinger V, Assmus B, Britten MB, Honold J, Lehmann R, Teupe C, et al. 2004. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarctionfinal one-year results of the TOPCARE-AMI Trial. J Am Coll Cardiol. 44:1690–1699.
- Schaer DA, Murphy JT, Wolchok JD. 2012. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol. 24:217–224.
- Schreiber TH, Raez L, Rosenblatt JD, Podack ER. 2010. Tumor immunogenicity and responsiveness to cancer vaccine therapy: the state of the art. Semin Immunol. 22:105–112.
- Schulenburg A, Herrmann H, Karlic H, Mirkina I, Hubmann R, Laffer S, et al. 2010. Neoplastic stem cells: current concepts and clinical perspectives. Crit Rev Oncol Hematol. 76:79–98.
- Scott AM, Allison JP, Wolchok JD. 2012. Monoclonal antibodies in cancer therapy. Cancer Immun. 12:14.
- Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP. 2008. Epitope landscape in breast and colorectal cancer. Cancer Res. 68:889–892.
- Smith GP. 1985. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 228:1315–1317.
- Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, et al. 2012. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. 379:39–46.
- Sobol RE. 2006. The rationale for prophylactic cancer vaccines and need for a paradigm shift. Cancer Gene Ther. 13:725–731.
- Srivastava PK. 2005. Immunotherapy for human cancer using heat shock protein-peptide complexes. Curr Oncol Rep. 7:104–108.
- Steinman RM, Banchereau J. 2007. Taking dendritic cells into medicine. Nature. 449:419–426.
- Stevenson FK. 2004. DNA vaccines and adjuvants. Immunol Rev. 199:5–8.
- Stevenson FK, Ottensmeier CH, Johnson P, Zhu D, Buchan SL, Mccann KJ, et al. 2004. DNA vaccines to attack cancer. Proc Natl Acad Sci U S A. 101 Suppl 2:14646–14652.
- Strand S, Galle PR. 1998. Immune evasion by tumours: involvement of the CD95 (APO-1/Fas) system and its clinical implications. Mol Med Today. 4:63–68.
- Strioga MM, Felzmann T, Powell DJ Jr., Ostapenko V, Dobrovolskiene NT, Matuskova M, et al. 2013. Therapeutic dendritic cell-based cancer vaccines: the state of the art. Crit Rev Immunol. 33:489–547.
- Tabi Z, Man S. 2006. Challenges for cancer vaccine development. Adv Drug Deliv Rev. 58:902–915.
- Tang DC, Devit M, Johnston SA. 1992. Genetic immunization is a simple method for eliciting an immune response. Nature. 356:152–154.
- Tarhini A, Lo E, Minor DR. 2010. Releasing the brake on the immune system: ipilimumab in melanoma and other tumors. Cancer Biother Radiopharm. 25:601–613.
- Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. 2008. Anti-CD38 antibody–mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 112:568–575.
- Taylor DD, Taylor CG, Jiang CG, Black PH. 1988. Characterization of plasma membrane shedding from murine melanoma cells. Int J Cancer. 41:629–635.
- Teeling JL, French RR, Cragg MS, van Den Brakel J, Pluyter M, Huang H, et al. 2004. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 104:1793–1800.
- Theobald M, Biggs J, Dittmer D, Levine AJ, Sherman LA. 1995. Targeting p53 as a general tumor antigen. Proc Natl Acad Sci U S A. 92:11993–11997.
- Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, et al. 2002. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 99:1517–1526.
- Triozzi PL, Allen KO, Carlisle RR, Craig M, Lobuglio AF, Conry RM. 2005. Phase I study of the intratumoral administration of recombinant canarypox viruses expressing B7.1 and interleukin 12 in patients with metastatic melanoma. Clin Cancer Res. 11: 4168–4175.
- Vaklavas C, Forero-Torres A. 2012. Safety and efficacy of brentuximab vedotin in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Ther Adv Hematol. 3:209–225.
- Vazquez AM, Hernandez AM, Macias A, Montero E, Gomez DE, Alonso DF, et al. 2012. Racotumomab: an anti-idiotype vaccine related to N-glycolyl-containing gangliosides - preclinical and clinical data. Front Oncol. 2:150.
- Vinay DS, Kwon BS. 2012. Immunotherapy of cancer with 4–1BB. Mol Cancer Ther. 11:1062–1070.
- Vu T, Claret FX. 2012. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2:62.
- Whiteside TL. 2005. Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system? Br J Cancer. 92:209–211.
- Wollert KC, Meyer GP, Lotz J, Ringes-Lichtenberg S, Lippolt P, Breidenbach C, et al. 2004. Intracoronary autologous bone-marrow cell transfer after myocardial infarction: the BOOST randomised controlled clinical trial. Lancet. 364:141–148.
- Yaddanapudi K, Mitchell RA, Eaton JW. 2013. Cancer vaccines: Looking to the future. Oncoimmunology. 2:e23403.
- Yajima N, Yamanaka R, Mine T, Tsuchiya N, Homma J, Sano M, et al. 2005. Immunologic evaluation of personalized peptide vaccination for patients with advanced malignant glioma. Clin Cancer Res. 11:5900–5911.
- Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG, Jakobovits A. 1999. Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res. 59:1236–1243.
- Yee C. 2013. Adoptive T-cell therapy for cancer: boutique therapy or treatment modality? Clin Cancer Res. 19:4550–4552.
- Yu M, Finn OJ. 2006. DNA vaccines for cancer too. Cancer Immunol Immunother. 55:119–130.
- Zent CS, Secreto CR, Laplant BR, Bone ND, Call TG, Shanafelt TD, et al. 2008. Direct and complement dependent cytotoxicity in CLL cells from patients with high-risk early-intermediate stage chronic lymphocytic leukemia (CLL) treated with alemtuzumab and rituximab. Leuk Res. 32:1849–1856.
- Zhou G, Lu Z, Mccadden JD, Levitsky HI, Marson AL. 2004. Reciprocal changes in tumor antigenicity and antigen-specific T cell function during tumor progression. J Exp Med. 200:1581–1592.
- Zhou H, Luo Y, Lo JF, Kaplan CD, Mizutani M, Mizutani N, et al. 2005. DNA-based vaccines activate innate and adaptive antitumor immunity by engaging the NKG2D receptor. Proc Natl Acad Sci U S A. 102:10846–10851.
- Zou W. 2005. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 5:263–274.
- zum Buschenfelde CM, Hermann C, Schmidt B, Peschel C, Bernhard H. 2002. Antihuman epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances cytolytic activity of class I-restricted HER2-specific T lymphocytes against HER2-overexpressing tumor cells. Cancer Res. 62:2244–2247.