
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The purpose of this study was to develop PEGylated nanoparticles of bendamustine (BM) to improve therapeutic efficiency of drug and reduce the side-effects. The nanoparticles were prepared by a modified diffusion–emulsification method. The particle size and zeta potential of optimized BM-loaded PEGylated NPs were found to be 256 nm and −29.1 mV. The in vitro release showed biphasic behavior, with initial burst release followed by slow sustained delivery. The anti-tumor activity was determined using the A- 549 cell line, by the MTT assay. The stability study revealed that the nanoparticles prepared were stable for 3 months at both 25°C and 4°C.
Introduction
Lung cancer is the most common non-skin cancer in the developed countries and the second most common overall, worldwide. According to WHO, it occurs mainly in men but is still the second most common cancer in women, with 1.3 million deaths worldwide annually (CitationWHO 2009). Basically, lung cancers, or bronchogenic carcinomas, are of two types, that is, small-cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC). NSCLC accounts for about 80% of all lung cancers (CitationTravis et al. 1995). Both NSCLC and SCLC may be treated with chemotherapy. Chemotherapeutic agents may be given alone as an adjuvant to surgical therapy, or in combination with radiotherapy. Although a number of chemotherapeutic drugs have been developed, the classes of drugs known as the platinum-based drugs have been the most effective in the treatment of lung cancers.
BM is a cytostatic agent used in the treatment of non-Hodgkin’s lymphomas. It exhibits bifunctional activity, that is, it combines DNA-alkylating properties with those of purine analogs. This feature contributes to its unique nature. It is also recommended for leukemia, sarcoma, and in lung cancer (CitationGidwani and Vyas 2014, CitationGandhi 2002). Although it is a very active alkylating agent, its pharmacological actions are hindered due to its poor solubility and bioavailability. It is administered parenterally, which causes severe adverse effects and dose-dependent side effects which lead to poor patient compliance. Presently, more than 80 clinical trials with BM are going on, which indicates that there is an increasing interest in the newer drug molecules rather than in older available drugs (CitationUS National Institutes of Health 2011).
PEGylation is the process of covalent attachment of polyethylene glycol polymer chains to another molecule. Polyethylene glycol (PEG) coating or conjugation is known as PEGylation. PEG is a nontoxic and nonirritant hydrophilic polymer (CitationJiang et al. 2006). PEGylation is routinely achieved by incubation of a reactive derivative of PEG with the target macromolecule (CitationGao et al. 2007). The covalent attachment of PEG to a drug or therapeutic protein can increase the hydrodynamic size (size in solution) of the agent, which prolongs its circulatory time by reducing renal clearance. PEGylation can also provide water solubility to hydrophobic drugs, improved drug solubility, reduced dosage frequency without diminished efficacy, with potentially reduced toxicity, extended circulating life, increased drug stability, and enhanced protection from proteolytic degradation (CitationTsutsumi et al. 1996).
Numerous approaches have been utilized to overcome the limitations or issues that hinder the therapeutic activity of BM. Entrapment of anticancer drugs in colloidal carriers can control their cell and tissue distribution profiles (CitationDanhier et al. 2009). Moreover, PEGylation of nanoparticles has been proven to be an effective approach for extending circulation in the blood stream, owing to the steric hindrance of the PEG chains (CitationShibata et al. 2004). Similarly, PEGylated polymers may improve the stability of the drug delivery system in the blood by preventing protein absorption and uptake by reticulo-endothelial systems (RES) (CitationTerada et al. 2007). PEGylated nanoparticles are used for tumor targeting. Attachment of ligand with the PEGylated nanoparticles is the most recent technique for cancer targeting (CitationGupta et al. 2015, CitationChen et al. 2011).
The aim of the present study was to prepare polymeric PEGylated solid lipid nanoparticles of BM for effective treatment of lung cancer. This approach will improve the therapeutic activity of BM, and also achieve a reduction of side effects.
Materials and method
Bendamustine (BM) was obtained as a gift sample from Dr. Reddy's Laboratory, Hyderabad (India). Palmitic acid, stearic acid, PEG 6000, Tween 80, and Span 80 were purchased from SDFCL Ltd., Mumbai. All other reagents and chemicals used in the study were of analytical grade.
Experimental work
Preparation of PEGylated solid lipid nanoparticles
The nanoparticles were prepared by the diffusion–emulsification method (CitationMu and Feng 2003). Briefly, PVA (emulsifier) (approx. 15 mg) was dissolved in hot water (500 ml) and heated at 80°C. To this surfactant solution, drug (BM, approx. 50 mg) and PEG 6000 (7.5 mg) were added. Lipid (palmitic acid) (150 mg) was melted at 75°C. The melted lipid solution was then added to aqueous solution and stirred continuously at 100 rpm for 30 min at 75°C. The melted mixture was then dispersed in 5% glucose solution, under mechanical agitation, to achieve a final emulsion of SLNs. The emulsion was further sonicated for 5 min, and finally, to remove the unencapsulated drug, the dispersion was filtered (1.2 μm) and ultracentrifuged at 12, 000 rpm for 20 min at 4°C.
Determination of BM content in PEGylated SLNs
The entrapment efficiency (EE) and drug loading (DL) of SLNs were estimated by determining the amount of free drug (BM) in the aqueous solution, which was separated by using the cooling centrifuge (Remi Instruments Ltd., Mumbai, India). For this, the dispersion of SLNs was centrifuged at 12,000 rpm for 20 min at 4°C. The concentration of BM in the aqueous phase was determined using a UV–visible spectrophotometer (UV 1700, Shimadzu, Japan) at λ max of 232 nm. The %EE and %DL were calculated by the following equations:
Measurement of particle size and zeta potential
The mean particle size and zeta potential of the SLNs were determined in purified water by the dynamic light scattering method using the Malvern Zetasizer ZS 90, USA. The zeta potential was calculated from the electrophoretic mobility using the Smoluchowski equation.
where, ζ is zeta potential, EM is the elecrophoretic mobility, η is the viscosity of the dispersion medium, and ε is the dielectric constant (CitationManjunath and Venkateswarlu 2005).
Determination of in vitro drug release from PEGylated SLNs
In vitro release of BM from SLNs was determined by the modified Franz diffusion method. A dialysis membrane (MW cutoff 10,000 Da) was used for the study. Prior to the study, this membrane was kept in distilled water for 24 h. The SLN dispersion (approx. 2 ml) was placed in the donor compartment, and the receptor compartment was filled with dissolution medium (pH 6.8 phosphate buffer) maintained at 35 ± 0.5°C with continuous stirring at 100 rpm. At regular time intervals, aliquots of samples were withdrawn from the receptor compartment and the exact volume of dissolution medium was added in the same compartment to maintain constant volume throughout the study. The amount of BM was estimated by UV–visible spectroscopy (CitationPaliwal et al. 2009).
In vitro cytotoxicity study
The cytotoxic activity of PEGylated nanoparticles of BM against A549 cells was evaluated using the MTT assay to quantify the survival rate. A-549 cells were added to each well of a 96-well plate and allowed to recover for 48 h. After recovery, free drug, blank nanoparticles, and drug-loaded PEGylated nanoparticles were added to the wells. Cells were treated for 48 h, and then 20 μL of MTT reagent (5 mg/mL) was added to the well. After incubation for 3 h, the plates were centrifuged and the supernatant was discarded. The formazan product was solubilized and the supernatant was separated from cell debris and remaining particles via centrifugation. Absorbance of the formazan solution was measured at 570 nm (650 nm background) on a Spectra Max plate reader. The results were expressed as mean ± standard deviation of 3 measurements (CitationTabatabaei Mirakabad et al. 2015, CitationLuo et al. 2014).
Antitumor activity
Tumor fragments obtained from different xenografts were transplanted (S.C.) in nude mice. When the volume of tumor reached approximately 250 mg, the mice were randomized for oral treatment with the drug BM at the dose of 2.5, 5, 10 or 20 mg/kg daily for one month. Each group consisted of 6 mice. The experiment was performed in two groups, namely, group I, treated with pure drug suspension, and group II, treated with PEGylated solid lipid nanoparticles of BM. The diameter of the tumor was measured three times a week, and the tumor volume was determined using the formula [l*h²/2]; (where l is the largest diameter of the tumor and h is the perpendicular axis). Data were expressed in terms of % of tumor growth inhibition, T/C % [(mean tumor weight of treated tumors/mean tumor weight of control tumors) × 100] (CitationPark et al. 2009).
In vivo pharmacokinetic studies
The in vivo pharmacokinetic study was performed on tumor-bearing mice (tumor weight approximately 400–450 mg) and were treated with PEGylated SLNs of BM at different doses of 2.5, 5, 10, and 20 mg/kg daily for 7 days. The samples of tumor and plasma were collected at 4 and 24 h after the last drug treatment. For sampling, mice were anesthetized and blood was collected from the retro-orbital plexus into heparinized tubes, and plasma was separated. The plasma and tumor levels were estimated.
Stability study
The storage stability study of PEGylated SLNs of BM was carried out according to ICH guidelines. For this, 10 ml of SLN dispersion with 2 mg/ml of drug concentration was stored at 4°C and 25°C for 3 months. Sampling was done every month, and the sample was analyzed for changes in particle size, zeta potential, and % EE.
Results and discussion
Particle size, zeta potential, and PDI of SLNs
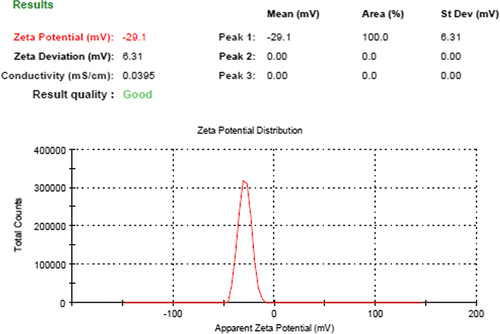
The particle size of optimized PEGylated SLNs was found to be 256.0 ± 1.13 nm; with PDI of 0.540. The size of blank nanoparticles (without drug) was less than that of the drug-loaded nanoparticles, that is, 216.38 ± 0.18 nm. The zeta potential of PEGylated SLNs was − 29.1 mV, which was also greater than the zeta potential of blank SLNs (− 21.23 mV). With the increase in concentration of BM, the size of the nanoparticles is gradually increased, but more than optimal dose of drug may cause aggregation and contribute to larger-sized particles with non-uniformity. The average particle size and zeta potential of PEGylated SLNs loaded with BM are shown in and respectively.
Figure 1. Particle size of PEGylated SLNs loaded with BM.
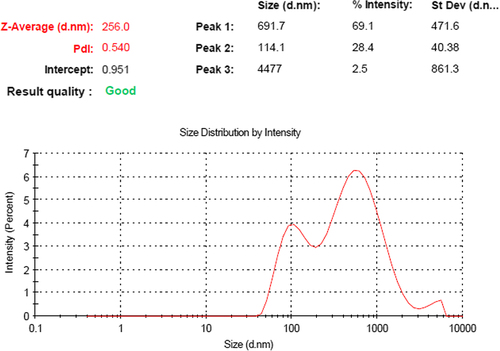
Figure 2. Zeta potential of PEGylated SLNs loaded with BM.
Percentage entrapment efficiency and drug-loading of PEGylated SLNs
High loading of BM—up to nearly 10% drug wt/wt—was achieved. The EE of PEGylated SLNs was found to be 77%. shows the data for physicochemical characterization (mean particle size, zeta potential, PDI, %EE and %DL) of SLNs.
Table I. Physicochemical characterization of PEGylated SLNs.
In vitro drug release study
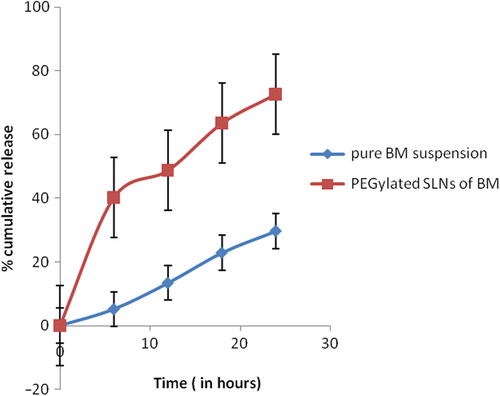
The release profile of BM-loaded PEGylated nanoparticles was investigated in PBS at 37°C. The cumulative percentage release is shown in . After the initial burst release for about 2 h, the release rate of BM slowed down and became almost zero-order. During the first 4 h, the release of BM was equivalent to 35.7 ± 2.11%. Furthermore, the amount of cumulative BM release was almost 72.6 ± 1.05%. The initial burst effect was due to the dissolution and diffusion of the drug that was poorly entrapped in the polymer matrix, while the slower and continuous release may be due to the diffusion of the drug localized in the PEGylated core of the nanoparticles.
Figure 3. Percentage cumulative drug release.
In vitro cytotoxicity study
The biological activity of PEGylated SLNs loaded with BM was determined against A-549 lung cancer cell line. Cytotoxicity of PEGylated nanoparticles was compared to that of the free drug (BM), non-drug-loaded nanoparticles, and with no treatment. PEGylated BM-loaded nanoparticles were more toxic than the free drug. A dose of 1 μM of free BM killed only 45% of the cell population after 48 h. However, in case of PEGylated BM SLNs, a dose of 1 μM killed more than 72% of the population. Blank nanoparticles did not influence/affect the cell viability in the culture. The IC50 value for PEGylated SLNs loaded with BM was 13.27 ± 0.62 nM after incubation for 48 h, more effective than pure drug (). The comparative analysis is shown in .
Figure 4. Cytotoxicity assay of BM in the A-549 lung cancer cell line.
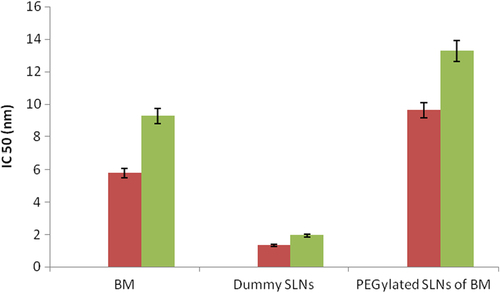
Table II. Cytotoxicity MTT assay of SLNs in the A-549 lung cancer cell line.
Antitumor activity
The in vivo antitumor effects of BM against A-549 human lung tumor were investigated by comparing with control, free drug (BM), and PEGylated nanoparticles of BM. The tumor growth was slowest in a group of animals treated with PEGylated SLNs of BM. Also, a significant difference in the inhibition rate of tumor mass was observed between the different study groups. The tumor volume increased with control, free BM, and dummy PEGylated SLN administration. However, there was no change in tumor volume in animals treated with PEGylated SLNs loaded with BM. shows the inhibition efficiency in different treatment groups. The average size and volume of the tumor, measured three times a week, is shown in . Data indicates that the size and volume of tumor was maximum in the morning and decreased slowly during the afternoon and night. The best effects were observed during the night, as during sleep, the tumor cells are not active, and therefore a prominent reduction in size and volume was seen. Moreover, there was very little improvement in first week, but as time passed, and at the end of the third week, maximum recovery was observed. These results prove that PEGylated nanoparticles of BM led to significant reduction in tumor mass and volume in 28 days. Results are shown as an average of three experimental readings and expressed as mean ± standard deviation. The changes in tumor size and volume during 28 days at different time intervals are shown in and .
Figure 5. Changes in tumor size during 28 days at different time intervals.
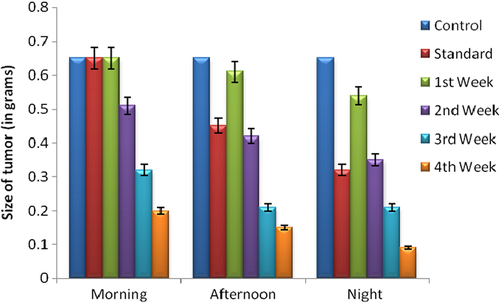
Figure 6. Changes in tumor volume during 28 days at different time intervals.
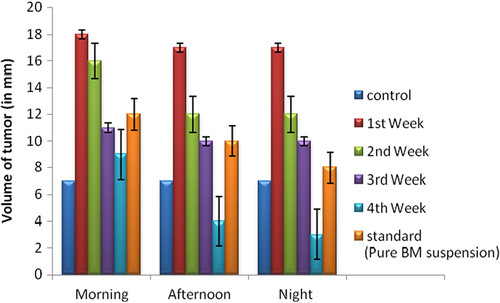
Table III. Tumor mass and inhibition efficiency after treatment (Mean ± SD) (n = 5).
Table IV. Average size and volume of tumor during 21 days of treatment with PEGylated NPs of BM.
In vivo pharmacokinetic study
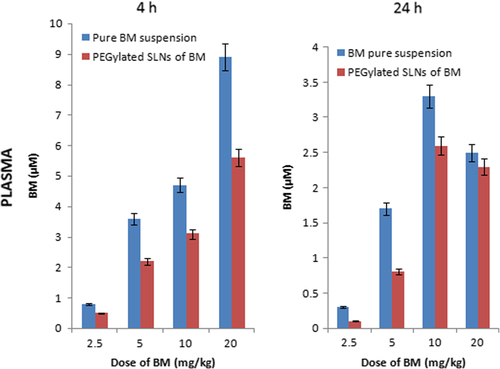
The concentration of BM was in the micro/sub-micro molar range at 4 and 24 h after the last administration of a 7-day schedule. The amount of drug accumulated in tumor after repeated treatment of PEGylated SLNs of BM for tumor/plasma ratios at 4 and 24 h were 2.5 ± 0.11and 12.6 ± 1.15 respectively. Both pure suspension of BM and PEGylated nanoparticles of BM were administered orally. After 2 h, the drug level in plasma and tumor increased suddenly due to burst effect. This may be due to PEGylation. An initial fast burst effect led to significant increase in the level of BM in plasma and tumor, followed by a slow sustained effect for 24 h. The PEGylation and nanosize of the particles synergistically led to effective release of BM. The comparative data of pharmacokinetic responses in plasma and tumor are shown in and .
Figure 7. Tumor levels of BM at 4 and 24 h after drug treatment.
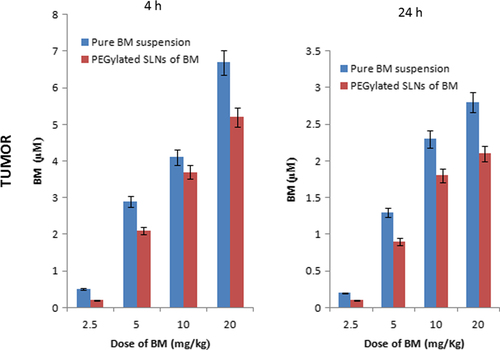
Figure 8. Plasma levels of BM at 4 and 24 h after drug treatment.
Stability study
PEGylated SLNs loaded with BM were found to be stable for 3 months at both the temperatures (4°C and 25°C), with slight variation in particle size, zeta potential, and EE. The results are shown in .
Table V. Stability studies of PEGylated SLNs.
Discussion
Development of a PEGylated nano-based system provides a convenient method for delivery of drugs to the lungs. Through PEGylation, more intact drug can be delivered at the respective site, as compared to free drug. PEGylation is a promising way to overcome the issue of EPR effect. It increases the circulation time of nanoparticles, thereby providing targeted delivery to specific organs. Moreover, it helps in improving the efficacy of the drug by assisting its delivery in lungs through biodistribution. PEGylated nanoparticles in the size range of 50–500 nm reduce the opsonization, increase the physiological stability, and enhance the possibility of reaching tumor cells. The physicochemical parameters (particle size, zeta potential, PDI, and surface morphology) play a vital role in controlling the biofunctionalization of the delivery system.
The particle size plays an important role in improving the bioavailability of the drug-loaded NPs. After PEGylation, the particle size is increased and the zeta potential is improved. Smaller sized particles tend to accumulate in the lesion sites due to facilitated extravasation, and hence, greater internalization occurs. Moreover, smaller particles possess higher zeta potential value, as compared to larger NPs. This is so because the small-sized NPs have a higher velocity of migration in a known applied electric field, and hence have a higher zeta potential value, resulting in increased stability of NPs in colloidal dispersions. Such particles are best suited for prolonged drug circulation and avoid rapid clearance and uptake by the RES system. The in vitro release profile of the PEGylated system prominently shows biphasic behavior (an initial burst release and slow sustained release later). The rapid initial burst drug release observed in the present study is commonly seen in the polymeric NPs (CitationZhang et al. 2004). These NPs are biocompatible, and thus do not cause any significant toxicity in vitro. The reason for high initial burst release may be the crystal structure of BM being transformed from a crystalline to an amorphous structure due to PEGylation. Moreover, low aqueous solubility may be a reason for the slow release of BM from the polymer matrices after burst release. Higher targeted efficiency of PEGylated SLNs loaded with BM was observed with A-549 cells, both in vitro and in vivo. The results of the in vitro cytotoxicity studies indicate that all tested drug-loaded nanoparticles significantly reduced IC50 values in the lung cell line, over the free drug.
The overall results demonstrated higher site-specific delivery, targeted efficiency, sustained delivery, improved cytotoxicity, and enhanced antitumor activity of PEGylated NPs of BM for effective treatment of lung cancer.
Conclusion
In the present work, we developed economical and safe BM-loaded PEGylated nanoparticles for efficient drug delivery for treatment of lung cancer. This approach will serve as a unique platform for site-specific and sustained delivery of BM with enhanced anticancer activity. PEGylated nanoparticles loaded with BM showed a favorable and prominent drug distribution profile in the tumor models. The in vivo study suggested that the PEGylated nanoparticles are more active as compared to pure drug. Thus, our findings support the potential use of PEGylated nanoparticles for clinical treatment of human lung cancer.
Acknowledgment
The authors are thankful to the Columbia Institute of Pharmacy for providing the necessary infrastructure for the completion of this work. The authors acknowledge and thank the Department of Pharmaceutical Analysis and Quality Assurance for providing the financial assistance and support for this work.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Chen J, Li S, Shen Q, He H, Zhang Y. 2011. Enhanced cellular uptake of folic acid-conjugated PLGA-PEG nanoparticles loaded with vincristine sulfate in human breast cancer. Drug Dev Ind Pharm. 37:1339–1346.
- Danhier F, Lecouturier N, Vroman B, Jerome C, Marchand-Brynaert J, Feron O, Preat V. 2009. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: In vitro and in vivo evaluation. J Control Release 133:11–17.
- Gandhi V. 2002. Metabolism of mechanism of action of bendamustine rationales for combination therapies. Semin Oncol. (4 Supp 13): 4–11.
- Gao JQ, Eto Y, Yoshioka Y, Sekiguchi F, Kurachi S, Morishige T. 2007. Effective tumor targeted gene transfer using PEGylated adenovirus vector via systemic administration. J Control Release. 122:102–110.
- Gidwani B, Vyas A. 2014. Formulation, characterization and evaluation of cyclodextrin-complexed bendamustine-encapsulated PLGA nanospheres for sustained delivery in cancer treatment. Pharm Dev Technol. 1–11.
- Gupta A, Kaur CD, Saraf S, Saraf S. 2015. Formulation, characterization, and evaluation of ligand-conjugated biodegradable quercetin nanoparticles for active targeting. Artif Cells Nanomed Biotechnol. (doi:https://doi.org/10.3109/21691401.2015.1008503).
- Jiang X, Dai H, Leong KW, Goh SH, Mao HQ, Yang YY. 2006. Chitosan-g-PEG/DNA complexes deliver gene to the rat liver via intrabiliary and intraportal infusions. J Gene Med. 8:477–487.
- Luo F, Li Y, Jia M, Cui F, Wu H, Yu F, Lin J, Yang X. 2014. Validation of a Janus role of methotrexate-based PEGylated chitosan nanoparticles in vitro. Nanoscale Res Lett. 9:363.
- Manjunath K, Venkateswarlu V. 2005. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Control Release. 107:215–228.
- Mu L, Feng S.S. 2003. A novel controlled release formulation for the anticancer drug Paclitaxel (Taxol): PLGA nanoparticles containing vitamin E TPGS. J Control Release. 86:33–48.
- Paliwal R, Rai S, Vaidya B, Khatri K, Goyal AK, Mishra N, et al. 2009. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 5:184–191.
- Park J, Fong PM, Lu J, Russell KS, Booth CJ, Saltzman WM, Fahmy TM. 2009. PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomedicine. 5:410–418.
- Shibata H, Yoshioka Y, Ikemizu S, Kobayashi K, Yamamoto Y. 2004. Functionalization of tumor necrosis factor-alpha using phage display technique and PEGylation improves its antitumor therapeutic window. Clin Cancer Res. 10:8293–8300.
- Tabatabaei Mirakabad FS, Akbarzadeh A, Milani M, Zarghami N, Taheri-Anganeh M, Zeighamian V, et al. 2015. A Comparison between the cytotoxic effects of pure curcumin and curcumin-loaded PLGA-PEG nanoparticles on the MCF-7 human breast cancer cell line. Artif Cell Nanomed Biotechnol. (doi: https://doi.org/10.3109/21691401.2014.955108).
- Terada T, Mizobata M, Kawakami S, Yamashita F, Hashida M. 2007. Optimization of tumor-selective targeting by basic fibroblast growth factor-binding peptide grafted PEGylated liposomes. J Control Release. 119:262–270.
- Travis WD, Travis LB, Devesa SS. 1995. Lung cancer incidence and survival by histological type. Lung Canc. 75:191–202.
- Tsutsumi Y, Tsunoda S, Kaneda Y, Kamada H, Kihira T, Nakagawa S, et al. 1996. In vivo anti-tumor efficacy of polyethylene glycol- modified tumor necrosis factor-alpha against tumor necrosis factor-resistant tumors. Jpn J Canc Res. 87:1078–1085.
- United States, National Institutes of Health, Cancer Therapy Evaluation Program (ctep). 2011. Common Toxicity Criteria (CTC).
- WHO, Disease and injury country estimates. 2009. WHO fact sheet on cancer.
- Zhang L, Hu Y, Jiang X, Yang C, Lu W, Yang YH. 2004. Camptothecin derivative loaded poly(caprolactone-co-lactide)-b-PEG-b-poly(caprolactone-co-lactide) nanoparticles and their biodistribution in mice. J Control Release. 96:135–148.