Abstract
The term “mitochondrial permeability transition” (MPT) refers to an abrupt increase in the permeability of the inner mitochondrial membrane to low molecular weight solutes. Due to osmotic forces, MPT is paralleled by a massive influx of water into the mitochondrial matrix, eventually leading to the structural collapse of the organelle. Thus, MPT can initiate mitochondrial outer membrane permeabilization (MOMP), promoting the activation of the apoptotic caspase cascade as well as of caspase-independent cell death mechanisms. MPT appears to be mediated by the opening of the so-called “permeability transition pore complex” (PTPC), a poorly characterized and versatile supramolecular entity assembled at the junctions between the inner and outer mitochondrial membranes. In spite of considerable experimental efforts, the precise molecular composition of the PTPC remains obscure and only one of its constituents, cyclophilin D (CYPD), has been ascribed with a crucial role in the regulation of cell death. Conversely, the results of genetic experiments indicate that other major components of the PTPC, such as voltage-dependent anion channel (VDAC) and adenine nucleotide translocase (ANT), are dispensable for MPT-driven MOMP. Here, we demonstrate that the c subunit of the FO ATP synthase is required for MPT, mitochondrial fragmentation and cell death as induced by cytosolic calcium overload and oxidative stress in both glycolytic and respiratory cell models. Our results strongly suggest that, similar to CYPD, the c subunit of the FO ATP synthase constitutes a critical component of the PTPC.
Introduction
Mitochondrial outer membrane permeabilization (MOMP) is widely considered as a point-of-no-return in the cascade of events whereby lethal signals that originate in the intracellular microenvironment are translated into apoptotic cell death.Citation1-Citation3 In addition, MOMP is involved in some instances of apoptosis as elicited by extracellular signals of distress as well as in non-apoptotic forms of cell death, including regulated necrosis.Citation4,Citation5 Irrespective of the initiating mechanism, MOMP results in: (1) the dissipation of the mitochondrial transmembrane potential (ΔΨm), in turn engendering a complete arrest of mitochondrial ATP synthesis and other ΔΨm-dependent activities (e.g., protein import) and (2) the release (into the cytosol) of cytotoxic proteins that normally serve physiological functions in the mitochondrial intermembrane space, such as cytochrome c.Citation6-Citation8 Thus, when MOMP affects the vast majority of a cell’s mitochondria both caspase-dependent and -independent mechanisms are activated to ultimately seal the cell’s fate.Citation9 Owing to its central role in cell death regulation, MOMP is involved in the pathophysiology of human diseases as diverse as cancer, ischemia and viral infections.Citation10,Citation11
MOMP can either be initiated at the outer mitochondrial membrane, owing to the pore-forming activity of the pro-apoptotic BCL-2 family members BAX and BAK,Citation12 or ensue the so-called “mitochondrial permeability transition” (MPT), an abrupt increase in the permeability of the inner mitochondrial membrane to low molecular weight solutes.Citation9,Citation13-Citation15 Due to osmotic forces, MPT results in a massive influx of water into the mitochondrial matrix, eventually leading to the structural breakdown of the organelle.Citation9,Citation13 MPT appears to be mediated by the so-called “permeability transition pore complex” (PTPC), a multiprotein entity lined up at the junctions between the inner and outer mitochondrial membranes.Citation9,Citation13 During the last couple of decades, two models have been put forward to mechanistically link MPT with the observation that the PTPC can assume two distinct states of conductance.Citation16 On one hand, it has been proposed that one of the main PTPC components, i.e., voltage-dependent anion channel (VDAC), would normally exist in an “open” state and would promote MPT upon “closure.”Citation17,Citation18 On the other hand, a low-conductance conformation of the PTPC has been suggested to allow for the physiological exchange of metabolites between the mitochondrial matrix and the cytosol. According to this model, the PTPC would mediate MPT following a conformational shift to a high-conductance state.Citation19,Citation20 To date, there is no model that would reconcile these two mutually exclusive working hypotheses.
The precise molecular composition of the PTPC has been subject of intense investigation yet remains poorly understood.Citation9 Indeed, whereas a few proteins have been indicated as core components of the PTPC, including voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT) and cyclophilin D (CYPD), genetic experiments have confirmed a critical contribution to MPT in vivo only for the latter.Citation21-Citation23 Conversely, neither VDAC nor any of the main isoforms of ANT appear to be required for the lethal functions of the PTPC.Citation24-Citation26 Along similar lines, dozens of cytosolic (e.g., hexokinase II) and mitochondrial (e.g., translocator protein of 18 KDa) interactors of the PTPC have been described, suggesting that the PTPC constitutes a highly dynamic signaling entity,Citation27 yet none of these proteins seems to be required for MPT-driven MOMP. Of note, both pro- and anti-apoptotic BCL-2 family members, including BAX, BID, BCL-2 and BCL-XL,Citation18,Citation20,Citation28 have been shown to physically bind to—and hence modulate the function of—PTPC components, indicating that the molecular machineries mediating MPT-driven and primary MOMP do not operate in a mutually exclusive manner.
The mitochondrial F1FO ATP synthase has previously been shown to interact with ANT and the mitochondrial phosphate carrier (SLC25A3), both of which are involved in MPT,Citation29,Citation30 to form multiprotein complexes that catalyze the final steps of mitochondrial ATP synthesis also known as “ATP synthasomes.”Citation31 In addition, CYPD as well as two distinct anti-apoptotic members of the BCL-2 family, namely BCL-XL and MCL-1, have recently been reported to regulate mitochondrial ATP synthesis by physically interacting with the F1FO ATP synthase.Citation32-Citation35 Driven by these observations and by the facts that: (1) a selective inhibitor of the FO subunit of ATP synthase, i.e., oligomycin, is able to prevent cell death as induced by tumor necrosis factor α (TNFα),Citation36 the multi-kinase inhibitor staurosporineCitation37 or BAX overexpression;Citation38 and that (2) the activity of both the PTPC and F1FO ATP synthase is modulated by Mg2+ ions;Citation39,Citation40 we decided to investigate the role of FO ATP synthase subunits in MPT.
Here, we reveal the important finding that the c subunit of the FO ATP synthase is required for MPT-driven mitochondrial fragmentation and cell death triggered by cytosolic calcium overload and oxidative stress.
Results and Discussion
Genetic manipulations of F1FO ATP synthase subunits
The membrane-spanning component of human mitochondrial ATP synthase (best known as FO, reflecting the fact that it can be inhibited by oligomycin) is composed by no less than nine polypeptides (a, b, c, d, e, f, g, F6 and 8), while the soluble catalytic core of the complex (F1) consists of five different subunits (α, β, γ, δ and ε).Citation41,Citation42 Among the nine polypeptides building up FO, a, b and c display an elevated degree of evolutionary conservation (subunits d-g, F6 and 8 are found in mitochondrial, but not prokaryotic, ATP synthases) and are sufficient for the complex to translocate protons across lipid bilayers.Citation43,Citation44 While the a subunit is encoded by mitochondrial DNA, b and c are encoded by the nuclear genome.Citation45
It has previously been shown that ρ0 cells (which lack mitochondrial DNA) are equipped with a functional PTPC, de facto excluding a prominent role for the a subunit of the FO synthase in MPT.Citation46,Citation47 Moreover, (1) the c, but not the b, subunit has been ascribed with conductive properties;Citation48 and (2) a peptide displaying a consistent degree of similarity to the c subunit has been indicated as a putative regulator of the PTPC.Citation49 Therefore, we focused on the putative contribution of the c subunit of the FO ATP synthase to the lethal functions of the PTPC.
To understand whether and how the c subunit participates in MPT, we decided to genetically manipulate its expression levels in a cell model that heavily rely on glycolysis (rather than on mitochondria) for ATP synthesis, i.e., human cervical carcinoma HeLa cells.Citation50 This choice stemmed from the fact that the activity of the PTPC is modulated by various intermediate metabolites, including NADH, ATP, ADP and inorganic phosphate.Citation9 As the c subunit is coded by three distinct genes (ATP5G1, ATP5G2 and ATP5G3) that generate polypeptides bearing different mitochondrial import sequences but forming the very same mature protein,Citation45 we had to envision a depletion strategy based on a mix of three commercial validated small-interfering RNAs (siRNAs) (). In addition, we generated a construct for the overexpression of MYC-tagged ATP5G1 under the control of the cytomegalovirus (CMV) immediate early promoter. Upon transfection, the exogenous c subunit properly co-localized with the mitochondrial marker heat shock 60 kDa protein 1 (HSPD1, best known as HSP60), as determined by two-color 3D deconvolution immunofluorescence microscopy (). As a control, we used a commercial non-targeting siRNA as well as a validated siRNA specific for the α polypeptide of the F1 subunit (ATP5A1) (Fig. S1A).
Figure 1. Mitochondrial alterations ensuing the downregulation or overexpression of the c subunit of the FO ATP synthase in HeLa cells. (A and B) Human cervical carcinoma HeLa cells were either transfected with a control siRNA (siSCR) or a mix of siRNAs targeting ATP5G1, ATP5G2 and ATP5G3 (siATP5G) for 48 h (A) or, alternatively, subjected to mock transfection or transfected with a plasmid encoding MYC-tagged ATP5G1 for 24 h (A and B), and then processed for either the immunoblotting-assisted detection of the FO c subunit, the FO a subunit, the F1 α subunit, cytochrome c (Cyt. c) and voltage-dependent anion channel 1 (VDAC1) (A) or the immunofluorescence-microscopy assisted visualization of heat shock 60 kDa protein 1 (HSPD1) and the FO c subunit (via the MYC tag). In (A) (reporting representative results), β actin levels were monitored to ensure the equal loading of lanes. (C and D) HeLa cells were transfected as in (A and B) but in combination with a plasmid coding for a mitochondrially targeted variant of luciferase, then stimulated with 100 μM histamine (Hist) and monitored for light emission over time upon the exogenous administration of luciferin. Representative traces as well as quantitative data illustrating the Hist-induced increase in luminescence (means ± SEM, n = 6) are reported. *p < 0.05 (unpaired Student’s t-test), as compared with equally stimulated, mock-transfected cells; n.s. = non-significant (unpaired Student’s t-test), as compared with equally stimulated, siSCR-transfected cells. (E) HeLa cells transfected as in (A and B) and then maintained in baseline conditions were stained with tetramethylrhodamine methyl ester (TMRM) for the assessment of mitochondrial transmembrane potential (ΔΨm). Quantitative data (means ± SEM, n = 5) are reported. *p < 0.05 (unpaired Student’s t-test), as compared with mock-transfected cells; n.s. = non-significant (unpaired Student’s t-test), as compared with siSCR-transfected cells. (F) HeLa cells were transfected as in (A and B) but in combination with a plasmid encoding a mitochondrially targeted variant of GFP, then maintained in control conditions and analyzed by 3D deconvolution fluorescence microscopy. Representative images and quantitative data illustrating the number of GFP+ 3D objects per cell (means ± SEM, n = 7) are reported. *p < 0.05 (unpaired Student’s t-test), as compared with mock-transfected cells; n.s. = non-significant (unpaired Student’s t-test), as compared with siSCR-transfected cells.
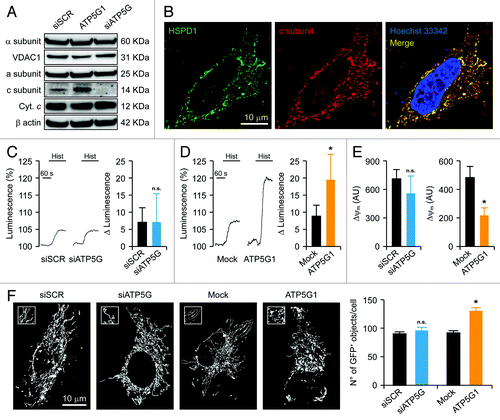
Histamine promotes the transfer of Ca2+ from reticular stores to mitochondria, resulting in increased mitochondrial ATP synthesis.Citation51,Citation52 The administration of 100 μM histamine to HeLa cells, however, had no effect on mitochondrial ATP levels (assessed with a mitochondrially targeted firefly luciferase) (). In addition, this response was not influenced by the depletion of the FO c subunit () or the F1 α subunit (Fig. S1B), confirming the near-to-purely glycolytic nature of this cellular model. Conversely, HeLa cells overexpressing the c subunit responded to histamine with a comparatively higher increase in mitochondrial ATP synthesis (). Such a metabolic effect was paralleled by a reduction in Δψm, as monitored in cellula by means of a Δψm-sensitive fluorochrome (i.e., tetramethylrhodamine methyl ester, TMRM) (). Moreover, the transfection-enforced overexpression of the FO c subunit promoted mitochondrial fragmentation, as assessed by the virtual reconstruction of the mitochondrial network and automated image analysis in cells co-expressing a mitochondrially targeted green fluorescent protein variant (mtGFP) (). Of note, the increased number of mtGFP+ objects observed in HeLa cells overexpressing the FO c subunit did not stem from a net increase in mitochondrial mass, as the protein levels of the FO a subunit, the F1 α subunit, cytochrome c and VDAC1 were not altered in this setting (). Moreover, neither Δψm nor the integrity of the mitochondrial network was significantly affected by the depletion of the FO c subunit () or that of the F1 α subunit (Fig. S1C).
Altogether, these results suggest that, in a near-to-purely glycolytic cell model, the genetic inhibition of F1FO ATP synthase subunits do not affect mitochondrial ATP synthesis.
Impact of the FO ATP synthase c subunit on MPT
To monitor the opening of the PTPC in living cells, we took advantage of the calcein-Co2+ assay.Citation53,Citation54 In this setting, cells are loaded with an esterified form of calcein, which is retained within the plasma membrane (thanks to the activity of cytoplasmic esterases) and freely diffuses into all subcellular compartments (including mitochondria), followed by bivalent cobalt (Co2+) ions. Co2+ ions quench the calcein-dependent cytoplasmic and nuclear, but not mitochondrial, fluorescence, as they are excluded from the mitochondrial matrix owing to the barrier function of the inner mitochondrial membrane. Thus, the induction of MPT can be followed in cellula as a further drop in calcein-dependent fluorescence.Citation53,Citation54
HeLa cells transfected with a control siRNA (or mock-transfected) and loaded with calcein-Co2+ responded to the administration of 1 μM ionomycin (a ionophore that triggers MPT by promoting a mitochondrial Ca2+ overload) with a rapid decrease in calcein fluorescence. Such a drop could be fully prevented by the pre-administration of cyclosporine a (CsA, a chemical inhibitor of CYPD), confirming that it resulted from the opening of the PTPC (). Importantly, the depletion of the FO c subunit (), but not that of the F1 α subunit (Fig. S1D), inhibited MPT as efficiently as did the pre-administration of CsA. In contrast, HeLa cells overexpressing the FO c subunit underwent MPT in response to 1 μM ionomycin with a consistently accelerated kinetics ().
Figure 2. Impact of the c subunit of the FO ATP synthase on MPT and MPT-driven mitochondrial fragmentation in HeLa cells. (A and B) Human cervical carcinoma HeLa cells were either transfected with a control siRNA (siSCR) or a mix of siRNAs targeting ATP5G1, ATP5G2 and ATP5G3 (siATP5G) for 48 h (A) or, alternatively, subjected to mock transfection or transfected with a plasmid encoding MYC-tagged ATP5G1 for 24 h (B), then loaded with calcein acetoxymethyl ester plus Co2+, optionally administered with 1 μM cyclosporine A (CsA), and stimulated with 1 μM ionomycin (Iono), followed by the fluorescence microscopy-assisted assessment of the calcein signal over time. Representative traces upon normalization to the initial calcein signal and quantitative data illustrating the calcein quenching rate (means ± SEM, n = 5–7) are reported. *p < 0.05 (unpaired Student’s t-test), as compared with siSCR- (A) or mock-transfected (B) cells. (C and D) HeLa cells were transfected as in (A and B) but in combination with a plasmid encoding a mitochondrially targeted variant of GFP, optionally administered with 1 μM CsA, and stimulated with 1 μM Iono, followed by the fluorescence microscopy-assisted monitoring of the GFP signal over time. Representative images and quantitative data illustrating the number of GFP+ 3D objects per cell at the indicated time after the administration of Iono (means ± SEM, n = 4) are reported. *p < 0.05 (unpaired Student’s t-test), as compared with siSCR- (C) or mock-transfected (D) cells receiving no CsA. #p < 0.05 (unpaired Student’s t-test), as compared with ATP5G1-overexpressing cells receiving no CsA (D).
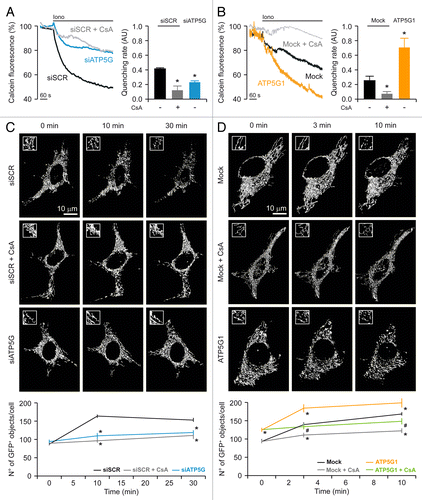
MPT is known to result in the structural collapse of the mitochondrial network, (at least in part) owing to the colloidosmotic swelling of the mitochondrial matrix.Citation9,Citation13 In line with this notion, HeLa cells transfected with a control siRNA and with a mtGFP-coding construct responded to 1 μM ionomycin by undergoing mitochondrial fragmentation, as indicated by a consistent increase in the number of mtGFP+ objects per cell. Such an increase could be virtually abolished by the pre-administration of CsA as well as by the depletion of the FO c subunit (), but not by that of the F1 α subunit (Fig. S1C). Conversely, mitochondrial fragmentation ensuing the opening of the PTPC in response to 1 μM ionomycin was largely aggravated by the overexpression of the c subunit of the FO ATP synthase, yet this phenomenon could be fully suppressed by CsA ().
Taken together, these findings demonstrate that the c subunit of the FO ATP synthase plays a critical role in the CsA-dependent opening of the PTPC induced by cytosolic Ca2+ overload.
Impact of the c subunit of the FO ATP synthase on MPT-driven MOMP and cell death
Together with the massive transfer of Ca2+ from the endoplasmic reticulum to the cytosol, oxidative stress, i.e., the condition in which reactive oxygen species (ROS) accumulate in spite of enzymatic and non-enzymatic antioxidant defense systems, is one of the best-characterized homeostatic perturbations that promotes PTPC opening in vitro and in vivo.Citation9,Citation21,Citation23 Accordingly, the mitochondria of HeLa cells transfected with a control siRNA and exposed to a high dose (2 mM) of the prototypic pro-oxidant hydrogen peroxide (H2O2) underwent a rapid and irreversible depolarization. Conversely, the ΔΨm of HeLa cells depleted of the FO c subunit dissipated upon the administration of high-dose H2O2 with a consistently delayed kinetics (). Along similar lines, while the mitochondria of mock-transfected HeLa cells responding to low-dose H2O2 (500 µM) depolarized in a relatively slow fashion, this process was dramatically accelerated in cells that overexpressed the c subunit of the FO ATP synthase (). Consistent with these observations, cytochrome c could be readily detected by immunoblotting in cytosolic extracts of HeLa cells transfected with a control siRNA and treated with H2O2 (), indicating that—in control conditions—these cells respond to oxidative stress by undergoing MOMP. Importantly, the amount of cytochrome c released into the cytosol by HeLa cells exposed to H2O2 was significantly reduced by the siRNA-mediated downregulation of the FO c subunit ().
Figure 3. Impact of the c subunit of the FO ATP synthase on MPT-driven MOMP and cell death in HeLa cells and rat cortical neurons. (A and B) Human cervical carcinoma HeLa cells were either transfected with a control siRNA (siSCR) or a mix of siRNAs targeting ATP5G1, ATP5G2 and ATP5G3 (siATP5G) for 48 h (A) or, alternatively, subjected to mock transfection or transfected with a plasmid encoding MYC-tagged ATP5G1 for 24 h (B), labeled with tetramethylrhodamine methyl ester (TMRM), and treated with the indicated concentration of hydrogen peroxide (H2O2), followed by the fluorescence microscopy-assisted assessment of mitochondrial transmembrane potential (Δψm) over time. One μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) was employed at the end of the assay to check for residual mitochondrial polarization. Representative traces upon normalization to the initial TMRM signal and quantitative data illustrating the calcein quenching rate (means ± SEM, n = 4–20) are reported. *p < 0.05 (unpaired Student’s t-test), as compared with equally treated, siSCR- (A) or mock-transfected (B) cells. (C) HeLa cells transfected as in (A) were treated with 1 mM H2O2 for 24 h, followed by the purification of cytosolic fractions and the immunoblotting-assisted detection of cytosolic cytochrome c (Cyt c). Representative results are reported. β actin levels were monitored to ensure the equal loading of lanes, while the presence of voltage-dependent anion channel 1 (VDAC) was assessed as an indicator of the purity of fractions. (D and E) HeLa cells transfected as in (A) were maintained in control conditions or administered with 10 μM ionomycin (Iono) or 1 mM H2O2 for 3 h, cultured in drug-free conditions for further 24 h, then labeled with propidium iodide (PI). Quantitative data illustrating the percentage of PI+ (dead) cells (means ± SEM, n = 3–6) are reported. *p < 0.05 (unpaired Student’s t-test), as compared with untreated, siSCR- (D) or mock-transfected (E) cells. #p < 0.05 (unpaired Student’s t-test), n.s. = non-significant (unpaired Student’s t-test), as compared with equally treated, siSCR- (D) or mock-transfected (E) cells. (F) Cortical neurons isolated from newborn rats were co-transfected with a plasmid coding for GFP and either siSCR or siATP5G for 48 h, optionally exposed to 500 μM glutamate for 30 min, cultured in drug-free conditions for further 24 h and eventually labeled with PI. Representative images and quantitative data illustrating the percentage of PI+ cells among GFP+ cell populations receiving glutamate (means ± SEM) are reported. Arrowheads indicate GFP+ cells. #p < 0.05 (unpaired Student’s t-test), as compared with siSCR-transfected, glutamate-treated neurons. (G) Rat cortical neurons transfected as in (F) were processed for the immunoblotting-assisted detection of the FO c subunit. Representative results are reported (β actin levels were monitored to ensure equal lane loading).
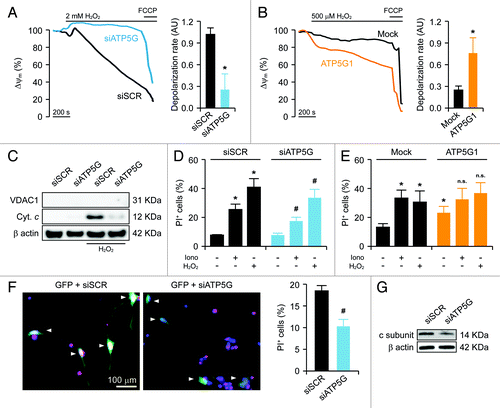
The ultimate functional consequence of MPT-driven MOMP is the activation of caspase-dependent and -independent executioner mechanisms of cell death.Citation4,Citation9 Indeed, approximately 25% and 40% of HeLa cells died upon exposure to ionomycin and H2O2, respectively, as indicated by the uptake of propidium iodide (PI), which can enter cells only upon the permeabilization of plasma membranes.Citation55 Importantly, the depletion of FO c subunit, but not that of the F1 α subunit, considerably reduced the lethal effect of Ca2+ overload and oxidative stress in HeLa cells (; Fig. S1E). The transfection-enforced overexpression of the FO c subunit per se induced some extent of cell death, yet failed to significantly aggravate the lethal response of HeLa cells to ionomycin and H2O2 (), reinforcing the notion that these genetic and pharmacological interventions modulate a single functional module.
Finally, to test our observations in a more physiologically relevant model of MPT-driven cell death, we monitored the response of cultured rat cortical neurons (which mainly rely on mitochondria for ATP synthesis) to glutamate, a neurotransmitter that causes the potentially lethal release of Ca2+ ions from the endoplasmic reticulum (a phenomenon known as excitotoxicity).Citation56,Citation57 To this aim, rat cortical neurons were transfected with a GFP-encoding plasmid (to focus the analysis on transfected cells) together with either a control siRNA or with a siRNA mix targeting the FO c subunit, followed by a short exposure (30 min) to glutamate 48 h later. Thereafter, neurons were cultured in standard conditions for additional 24 h and eventually labeled with PI. In these conditions, approximately 20% of GFP+ neurons transfected with the control siRNA stained positively for PI, yet only about 10% of neurons efficiently receiving the siRNAs specific for the FO c subunit did so (). Of note, the siRNAs originally designed to target the human form of the FO c subunit were efficient in cells from Rattus norvegicus as well, in spite of a few sequence misalignments ().
Altogether, these data demonstrate that the c subunit of the FO ATP synthase regulates not only MPT but also its major functional consequences, including MOMP and cell death, both in glycolytic and respiratory cell models.
Concluding remarks
As a pathophysiologically relevant process, MPT can precipitate the unwarranted demise of post-mitotic cells in response to multiple stimuli. In particular, MPT has been involved in the acute loss of neurons and cardiomyocytes induced by ischemia/re-oxygenation and excitotoxicity.Citation23,Citation58-Citation60 The molecular mechanisms that trigger the opening of the PTPC in these settings share a prominent oxidative component and the cytosolic accumulation of Ca2+ ions, which de facto are tightly linked to each other.Citation61 During the last decades, several proteins have been suggested to constitute core components of the PTPC, including various isoforms of VDAC and ANT as well as CYPD, yet only the latter appears to be truly required for MPT in vivo.Citation21,Citation22,Citation24-Citation26 Here, we demonstrate that the c subunit of the FO ATP synthase is also necessary for MPT and its functional consequences, i.e., MOMP and cell death.
It might be argued that our findings do not reflect a direct implication of the FO c subunit in MPT, but rather alterations in the levels of ATP, ADP and inorganic phosphate, since all these metabolites (and many others) are known to influence the propensity of the PTPC to open.Citation9 We are confident that this is not the case: since (1) the depletion of the FO c subunit in a near-to-purely glycolytic cell model, i.e., human cervical carcinoma HeLa cells,Citation50 failed to alter the levels of mitochondrial ATP synthesis, yet inhibited MPT, mitochondrial fragmentation and cell death as triggered by cytosolic Ca2+ overload or oxidative stress, and (2) the downregulation of the F1 α subunit (which—similar to the FO c subunit—is required for the catalytic activity of the F1FO ATP synthase)Citation62 completely failed to affect MPT and its functional consequences.
Experiments to elucidate the precise molecular mechanisms whereby the c subunit of the FO ATP synthase participates in MPT are underway. By ion exchange chromatography, we have succeeded in separating a multiprotein complex that contains ANT, VDAC1, CYPD, hexokinase, the FO c subunit as well as multiple F1 components (data not shown), in line with the existence of a supramolecular entity involving several constituents of the PTPC as well as the ATP synthasome.Citation27,Citation29-Citation31 Now, it will be interesting to identify the direct physical interactors of the FO c subunit among the core PTPC components and its best known regulators (including pro- and anti-apoptotic BCL-2 family members as well as the oncosuppressor protein TP53).Citation63,Citation64 To test the actual pathophysiological relevance of our findings, it will be crucial to generate appropriate knockout models. Most likely, similar to the knockout of genes coding for other factors involved in mitochondrial respiration (e.g., cytochrome c),Citation65 a whole body Atp5g1−/− genotype will turn out to be embryonic lethal. In addition, the murine genome appears to contain multiple Atp5g1-related coding sequences, further complicating the generation of bona fide Atp5g1−/− mice. The consistent degree of redundancy affecting PTPC-related proteins (many of which exist in at least three isoforms) has previously caused problems for the creation of Vdac- and Ant-deficient animals.Citation24-Citation26 Actually, the discovery of a fourth isoform of murine ANT (i.e., Ant4),Citation66 has casted doubts on the conclusions of Kokoszka and colleagues, indicating that ANT would be dispensable for MPT.Citation26 Irrespective of these unresolved issue, well-designed conditional knockout strategies and/or the identification of mutations by which the vital and lethal functions of the FO c subunit can be uncoupled may allow for the elucidation of exact contribution of this protein to MPT.
Finally, it is tempting to speculate, yet remains to be formally demonstrated, that agents targeting the FO c subunit and/or its interaction with other components of the PTPC might exert prominent cytoprotective effects by subverting MPT, MOMP and cell death as induced by pathogenic insults.
Materials and Methods
Chemicals, cell cultures and transfections
Unless otherwise noted, chemicals were purchased from Sigma-Aldrich, cell culture media and supplements from Gibco-Life Technologies and plasticware from Corning Life Sciences. Human cervical carcinoma HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin G sodium and 100 µg/mL streptomycin sulfate. Rat cortical neurons were obtained from 1–3-day-old rats, as previously described,Citation67 and maintained in GlutaMax®-containing Neurobasal®-A medium supplemented with B-27® supplements, 100 units/mL penicillin G sodium and 100 µg/mL streptomycin sulfate.
RNA interference experiments were performed by transfecting cells with a commercial control siRNA (AllStars RNAi Controls, herein referred to as siSCR), with a validated siRNA specific for ATP5A1 (siATP5A1, sense 5′-GGCUGGAUUUGAAGCUUAAdTdT-3′) or with a mix of siRNAs targeting ATP5G1 (5′-GCUCUGAUCCGCUGUUGUAdTdT-3′), ATP5G1 (5′-CGGAGAUACUGACAGAUGAdTdT-3′) and ATP5G3 (5′-AGGGCUCUACGGUAUUUAAdTdT-3′), all purchased from Qiagen. siRNAs were transfected by means of the HiPerfect® transfection reagent, as per manufacturer’s instructions.Citation68
For transient overexpression experiments, a pCMV6 entry-based plasmid coding for a MYC-tagged variant of ATP5G1 under the control of the CMV immediate early promoter (RC200292) was obtained from OriGene. For the quantification of mitochondrial ATP levels, a VR1012-based construct encoding a mitochondrially targeted variant of the Photinus pyralis luciferase under the control of the CMV immediate early promoter was employed.Citation50 For the study of mitochondrial morphology, a pcDNA3-based plasmid coding for a mitochondrially targeted variant of GFP (mtGFP) under the control of the CMV immediate early promoter was used.Citation52 For the quantification of cell death in neurons, a pcDNA3-based construct coding for GFP under the control of the CMV immediate early promoter was used.Citation69 Plasmid (co-)transfections were performed via the standard Ca2+-phosphate technique.Citation70
Quantification of cell death
Dead cells were quantified by virtue of their ability to take up propidium iodide (PI), reflecting the breakdown of the plasma membrane.Citation55,Citation71 To this aim, HeLa cells were processed as indicated in the Tali® Apoptosis Kit (Molecular Probes-Life Technologies) and analyzed on a Tali® Image-Based cytometer (Invitrogen-Life Technologies). Conversely, rat neurons were stained with 1 μM PI (Molecular Probes-Life Technologies) for 5 min at room temperature (RT), then fixed with 4% paraformaldehyde (PFA) and stained with 1 μM Hoechst 33342 (Molecular Probes-Life Technologies) for 20 min at RT. Coverslips were then mounted on slides and analyzed on an Axiovert 200M microscope (Carl Zeiss) equipped with a 40× water immersion objective (N.A. 1.2, from Carl Zeiss) and a CoolSnap HQ CCD camera (Photometrics). Fifty random fields per condition were acquired in MetaMorph® (Molecular Devices) and images were analyzed with CellProfiler (Broad Institute) using a customized pipeline for the quantification of PI+ GFP+ cells.
Calcein-Co2+ assays
HeLa Cells were loaded with 1 mM calcein acetoxymethyl ester and Co2+ as instructed by the Image-IT® LIVE Mitochondrial Transition Pore Assay Kit (Molecular Probes-Life Technologies). Cells were then imaged by means of 490 ± 20 nm excitation and 525 nm longpass emission filters on a Axiovert 200M fluorescence microscope equipped with a 40× water immersion objective (N.A. 1.2, from Carl Zeiss). Finally, images were analyzed with MetaMorph® and quenching rates were determined as the slopes of the fluorescence trace over a period of 60 sec post-stimulation.
Quantification of Δψm
Cells were loaded with 1 nM tetramethylrhodamine methyl ester (TMRM, from Molecular Probes-Life Technologies) in Krebs-Ringer buffer supplemented with 250 μM sulfinpyrazone, then placed in a humidified chamber at 37°C and imaged with a LiveScan Swept Field Confocal Microscope (Nikon Instruments Inc.) equipped with a 60× oil immersion (N.A. 1.4, from Nikon Instruments Inc.) every 30 sec for 30 min. TMRM fluorescence was analyzed by means of the NIS Elements software package (Nikon Instruments Inc.), and depolarization rates were defined as the slopes of the fluorescence trace over a period of 10 min post-stimulation.
Quantification of mitochondrial ATP
HeLa cells expressing a mitochondrially targeted variant of the Photinus pyralis luciferase were perfused with a modified Krebs-Ringer buffer containing 125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 1 mM CaCl2, 20 μM luciferin and 20 mM HEPES buffer (pH 7.4 at 37°C), and luciferin-dependent luminescence was monitored with a customized luminometer (Elettrofor), as previously described.Citation50
Analysis of mitochondrial morphology
HeLa cells expressing a mitochondrially targeted variant of GFP,Citation69 were imaged with an IX-81 automated epifluorescence microscope (Olympus) equipped with a 60× oil immersion objective (N.A. 1.35, from Olympus) and an ORCA-R2 CCD camera (Hamamatsu Photonics K.K.). Selected cells were followed over time, and z-stacks were subjected to digital deconvolution by means of a Wiener deconvolution filter and a theoretical point-spread function provided by the Xcellence software (Olympus). GFP+ objects were quantified with the “3D object counter” plug-in of the open-source Fiji software (freely available at http://fiji.sc/), whereas 3D representations were obtained with the “3D Viewer” plug-in.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed according to standard procedures.Citation72 Briefly, cells were fixed in 4% PFA for 20 min at RT, washed three times in PBS and permeabilized with 0.1% Triton X-100 (v:v in PBS) for 5 min at RT. Thereafter, unspecific binding sites were blocked by incubating cells in 2% powdered milk (w:v in PBS) for 1 h at RT. Cells were then incubated overnight at 4°C with primary antibodies specific for HSPD1 (1:100 in blocking buffer) or MYC (1:25 in blocking buffer) (both from Abcam). Finally, primary antibodies were revealed by means of appropriate AlexaFluor 488® and AlexaFluor 594® conjugates (Molecular Probes-Life Technologies). Images were acquired on an Axiovert 220 M microscope equipped with a 100× oil immersion Plan Neofluar® objective (N.A. 1.3, from Carl Zeiss) and a CoolSnap HQ CCD camera. Each field was acquired over 21 z-planes spaced by 0.5 μm, and z-stacks were deconvoluted with the “Parallel Iterative Deconvolution” plug-in of Fiji.
Immunoblotting
Immunoblotting was performed as previously described, with minor modifications.Citation73 In brief, to obtain whole-cell extracts, cells were washed, harvested and lysed in RIPA buffer (50 mM TRIS-HCl pH 7.8, 150 mM NaCl, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM dithiothreitol) supplemented with 2 mM Na3VO4, 2 mM NaF, 1 mM phenylmethylsulfonyl fluoride and Complete Protease Inhibitor Cocktail® (Roche Diagnostics Corp.). Alternatively, cytosolic extracts were obtained by digitonin permeabilization, as previously described.Citation74 Thereafter, protein extracts (30 µg/lane) were separated on precast 4–12% SDS-PAGE gels (Life Sciences), electrotransfered onto PVDF membranes (Bio-Rad) and probed with antibodies specific for the FO a subunit (Abcam), the FO c subunit (Abcam), the F1 α subunit (Abcam), cytochrome c (BD Biosciences) and VDAC1 (Abcam). An antibody specific for β actin (Sigma-Aldrich) was used to monitor equal lane loading. Finally, membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Southern Biotech), followed by chemiluminescence detection with the SuperSignal West Pico® reagent and CL-XPosure® X-ray films (both from Thermo Scientific-Pierce).
Statistical procedures
Unless otherwise indicated, assays were performed in triplicate independent instances, yielding comparable results. Data, which are presented as means ± SEM, were analyzed with Microsoft Excel (Microsoft Co.). Statistical significance was determined by means of ANOVA followed by two-tailed unpaired Student’s t-tests. p values < 0.05 were considered statistically significant.
| Abbreviations: | ||
| ANT | = | adenine nucleotide translocase |
| CMV | = | cytomegalovirus |
| CsA | = | cyclosporine A |
| CYPD | = | cyclophilin D |
| Δψm | = | mitochondrial transmembrane potential |
| HSPD1 | = | heat shock 60 kDa protein 1 |
| MOMP | = | mitochondrial outer membrane permeabilization |
| MPT | = | mitochondrial permeability transition |
| mtGFP | = | mitochondrially targeted green fluorescent protein |
| PFA | = | paraformaldehyde |
| PI | = | propidium iodide |
| RT | = | room temperature |
| siRNA | = | small-interfering RNA |
| TMRM | = | tetramethylrhodamine methyl ester |
| VDAC | = | voltage-dependent anion channel |
Additional material
Download Zip (373.6 KB)Acknowledgments
We would like to thank Dr. Massimo Negrini, Giulia Fonti, Augusto Bevilacqua and Oliver Kepp for their extraordinary help. A.R. and C.G. are financed by the Italian Ministry of Health and Associazione Italiana per la Ricerca sul Cancro (AIRC), respectively. A.B. is supported by a Fondazione Italiana Sclerosi Multipla (FISM) fellowship (2010/B/1). S.P. is supported by a FISM training fellowship (2010/B/13). S.M. is supported by Fondazione Italiana per la Ricerca sul Cancro (FIRC) fellowship. M.R.W. is supported by the Polish National Science Center (UMO-2011/11/M/NZ3/02128) and BIO-IMAGing in Research Innovation and Education (FP7-REGPOT-2010-1). G.K. is supported by the European Commission (ArtForce); Agence National de la Recherche (ANR); Ligue contre le Cancer (Equipe labelisée); Fondation pour la Recherche Médicale (FRM); Institut National du Cancer (INCa); LabEx Immuno-Oncologie; Fondation de France; Fondation Bettencourt-Schueller; AXA Chair for Longevity Research; Cancéropôle Ile-de-France and Paris Alliance of Cancer Research Institutes (PACRI). P.P. is financed by AIRC, Telethon (GGP09128 and GGP11139B), the Italian Ministry of Education, University and Research and the Italian Ministry of Health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/cc/article/23599
References
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11:621 - 32; http://dx.doi.org/10.1038/nrm2952; PMID: 20683470
- Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 2012; 13:780 - 8; http://dx.doi.org/10.1038/nrm3479; PMID: 23175281
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al, Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16:3 - 11; http://dx.doi.org/10.1038/cdd.2008.150; PMID: 18846107
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107 - 20; http://dx.doi.org/10.1038/cdd.2011.96; PMID: 21760595
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11:700 - 14; http://dx.doi.org/10.1038/nrm2970; PMID: 20823910
- Galluzzi L, Joza N, Tasdemir E, Maiuri MC, Hengartner M, Abrams JM, et al. No death without life: vital functions of apoptotic effectors. Cell Death Differ 2008; 15:1113 - 23; http://dx.doi.org/10.1038/cdd.2008.28; PMID: 18309324
- Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep 2012; 13:322 - 30; http://dx.doi.org/10.1038/embor.2012.19; PMID: 22402666
- Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 2006; 13:1423 - 33; http://dx.doi.org/10.1038/sj.cdd.4401950; PMID: 16676004
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87:99 - 163; http://dx.doi.org/10.1152/physrev.00013.2006; PMID: 17237344
- Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 2009; 10:481 - 94; http://dx.doi.org/10.1038/nrn2665; PMID: 19543220
- Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog 2008; 4:e1000018; http://dx.doi.org/10.1371/journal.ppat.1000018; PMID: 18516228
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292:727 - 30; http://dx.doi.org/10.1126/science.1059108; PMID: 11326099
- Brenner C, Grimm S. The permeability transition pore complex in cancer cell death. Oncogene 2006; 25:4744 - 56; http://dx.doi.org/10.1038/sj.onc.1209609; PMID: 16892087
- Di Lisa F, Carpi A, Giorgio V, Bernardi P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim Biophys Acta 2011; 1813:1316 - 22; http://dx.doi.org/10.1016/j.bbamcr.2011.01.031; PMID: 21295622
- Halestrap AP. What is the mitochondrial permeability transition pore?. J Mol Cell Cardiol 2009; 46:821 - 31; http://dx.doi.org/10.1016/j.yjmcc.2009.02.021; PMID: 19265700
- Crompton M, Costi A. A heart mitochondrial Ca2(+)-dependent pore of possible relevance to re-perfusion-induced injury. Evidence that ADP facilitates pore interconversion between the closed and open states. Biochem J 1990; 266:33 - 9; PMID: 2106875
- Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997; 91:627 - 37; http://dx.doi.org/10.1016/S0092-8674(00)80450-X; PMID: 9393856
- Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem 2001; 276:19414 - 9; http://dx.doi.org/10.1074/jbc.M101590200; PMID: 11259441
- Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A 2000; 97:3100 - 5; http://dx.doi.org/10.1073/pnas.97.7.3100; PMID: 10737788
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999; 399:483 - 7; http://dx.doi.org/10.1038/20959; PMID: 10365962
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434:658 - 62; http://dx.doi.org/10.1038/nature03434; PMID: 15800627
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 2005; 280:18558 - 61; http://dx.doi.org/10.1074/jbc.C500089200; PMID: 15792954
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005; 102:12005 - 10; http://dx.doi.org/10.1073/pnas.0505294102; PMID: 16103352
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 2007; 9:550 - 5; http://dx.doi.org/10.1038/ncb1575; PMID: 17417626
- Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol 2007; 9:487 - 9; http://dx.doi.org/10.1038/ncb0507-487; PMID: 17473857
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004; 427:461 - 5; http://dx.doi.org/10.1038/nature02229; PMID: 14749836
- Verrier F, Deniaud A, Lebras M, Métivier D, Kroemer G, Mignotte B, et al. Dynamic evolution of the adenine nucleotide translocase interactome during chemotherapy-induced apoptosis. Oncogene 2004; 23:8049 - 64; http://dx.doi.org/10.1038/sj.onc.1208001; PMID: 15377997
- Zamzami N, El Hamel C, Maisse C, Brenner C, Muñoz-Pinedo C, Belzacq AS, et al. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene 2000; 19:6342 - 50; http://dx.doi.org/10.1038/sj.onc.1204030; PMID: 11175349
- Alcalá S, Klee M, Fernández J, Fleischer A, Pimentel-Muiños FX. A high-throughput screening for mammalian cell death effectors identifies the mitochondrial phosphate carrier as a regulator of cytochrome c release. Oncogene 2008; 27:44 - 54; http://dx.doi.org/10.1038/sj.onc.1210600; PMID: 17621274
- Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem 2008; 283:26312 - 23; http://dx.doi.org/10.1074/jbc.M805235200; PMID: 18667415
- Ko YH, Delannoy M, Hullihen J, Chiu W, Pedersen PL. Mitochondrial ATP synthasome. Cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J Biol Chem 2003; 278:12305 - 9; http://dx.doi.org/10.1074/jbc.C200703200; PMID: 12560333
- Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol 2011; 13:1224 - 33; http://dx.doi.org/10.1038/ncb2330; PMID: 21926988
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 2012; 14:575 - 83; http://dx.doi.org/10.1038/ncb2488; PMID: 22544066
- Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, et al. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 2009; 284:33982 - 8; http://dx.doi.org/10.1074/jbc.M109.020115; PMID: 19801635
- Chinopoulos C, Konràd C, Kiss G, Metelkin E, Töröcsik B, Zhang SF, et al. Modulation of F0F1-ATP synthase activity by cyclophilin D regulates matrix adenine nucleotide levels. FEBS J 2011; 278:1112 - 25; http://dx.doi.org/10.1111/j.1742-4658.2011.08026.x; PMID: 21281446
- Shchepina LA, Pletjushkina OY, Avetisyan AV, Bakeeva LE, Fetisova EK, Izyumov DS, et al. Oligomycin, inhibitor of the F0 part of H+-ATP-synthase, suppresses the TNF-induced apoptosis. Oncogene 2002; 21:8149 - 57; http://dx.doi.org/10.1038/sj.onc.1206053; PMID: 12444550
- Santamaría G, Martínez-Diez M, Fabregat I, Cuezva JM. Efficient execution of cell death in non-glycolytic cells requires the generation of ROS controlled by the activity of mitochondrial H+-ATP synthase. Carcinogenesis 2006; 27:925 - 35; http://dx.doi.org/10.1093/carcin/bgi315; PMID: 16361271
- Matsuyama S, Xu Q, Velours J, Reed JC. The Mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol Cell 1998; 1:327 - 36; http://dx.doi.org/10.1016/S1097-2765(00)80033-7; PMID: 9660917
- Murataliev MB, Boyer PD. Interaction of mitochondrial F1-ATPase with trinitrophenyl derivatives of ATP and ADP. Participation of third catalytic site and role of Mg2+ in enzyme inactivation. J Biol Chem 1994; 269:15431 - 9; PMID: 8195184
- Novgorodov SA, Gudz TI, Brierley GP, Pfeiffer DR. Magnesium ion modulates the sensitivity of the mitochondrial permeability transition pore to cyclosporin A and ADP. Arch Biochem Biophys 1994; 311:219 - 28; http://dx.doi.org/10.1006/abbi.1994.1230; PMID: 8203884
- Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 1994; 370:621 - 8; http://dx.doi.org/10.1038/370621a0; PMID: 8065448
- Devenish RJ, Prescott M, Roucou X, Nagley P. Insights into ATP synthase assembly and function through the molecular genetic manipulation of subunits of the yeast mitochondrial enzyme complex. Biochim Biophys Acta 2000; 1458:428 - 42; http://dx.doi.org/10.1016/S0005-2728(00)00092-X; PMID: 10838056
- Greie JC, Deckers-Hebestreit G, Altendorf K. Secondary structure composition of reconstituted subunit b of the Escherichia coli ATP synthase. Eur J Biochem 2000; 267:3040 - 8; http://dx.doi.org/10.1046/j.1432-1033.2000.01327.x; PMID: 10806404
- Greie JC, Heitkamp T, Altendorf K. The transmembrane domain of subunit b of the Escherichia coli F1F(O) ATP synthase is sufficient for H(+)-translocating activity together with subunits a and c. Eur J Biochem 2004; 271:3036 - 42; http://dx.doi.org/10.1111/j.1432-1033.2004.04235.x; PMID: 15233800
- De Grassi A, Lanave C, Saccone C. Evolution of ATP synthase subunit c and cytochrome c gene families in selected Metazoan classes. Gene 2006; 371:224 - 33; http://dx.doi.org/10.1016/j.gene.2005.11.022; PMID: 16460889
- Marchetti P, Susin SA, Decaudin D, Gamen S, Castedo M, Hirsch T, et al. Apoptosis-associated derangement of mitochondrial function in cells lacking mitochondrial DNA. Cancer Res 1996; 56:2033 - 8; PMID: 8616847
- Masgras I, Rasola A, Bernardi P. Induction of the permeability transition pore in cells depleted of mitochondrial DNA. Biochim Biophys Acta 2012; 1817:1860 - 6; http://dx.doi.org/10.1016/j.bbabio.2012.02.022; PMID: 22402226
- McGeoch JE, Guidotti G. A 0.1-700 Hz current through a voltage-clamped pore: candidate protein for initiator of neural oscillations. Brain Res 1997; 766:188 - 94; http://dx.doi.org/10.1016/S0006-8993(97)00618-5; PMID: 9359602
- Azarashvili TS, Tyynelä J, Odinokova IV, Grigorjev PA, Baumann M, Evtodienko YV, et al. Phosphorylation of a peptide related to subunit c of the F0F1-ATPase/ATP synthase and relationship to permeability transition pore opening in mitochondria. J Bioenerg Biomembr 2002; 34:279 - 84; http://dx.doi.org/10.1023/A:1020204518513; PMID: 12392191
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A 1999; 96:13807 - 12; http://dx.doi.org/10.1073/pnas.96.24.13807; PMID: 10570154
- Pinton P, Pozzan T, Rizzuto R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J 1998; 17:5298 - 308; http://dx.doi.org/10.1093/emboj/17.18.5298; PMID: 9736609
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998; 280:1763 - 6; http://dx.doi.org/10.1126/science.280.5370.1763; PMID: 9624056
- Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis 2007; 12:803 - 13; http://dx.doi.org/10.1007/s10495-007-0720-1; PMID: 17294081
- Petronilli V, Miotto G, Canton M, Colonna R, Bernardi P, Di Lisa F. Imaging the mitochondrial permeability transition pore in intact cells. Biofactors 1998; 8:263 - 72; http://dx.doi.org/10.1002/biof.5520080314; PMID: 9914828
- Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10:221 - 37; http://dx.doi.org/10.1038/nrd3373; PMID: 21358741
- Nicholls DG, Budd SL, Castilho RF, Ward MW. Glutamate excitotoxicity and neuronal energy metabolism. Ann N Y Acad Sci 1999; 893:1 - 12; http://dx.doi.org/10.1111/j.1749-6632.1999.tb07813.x; PMID: 10672225
- Abramov AY, Duchen MR. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim Biophys Acta 2008; 1777:953 - 64; http://dx.doi.org/10.1016/j.bbabio.2008.04.017; PMID: 18471431
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012; 149:1536 - 48; http://dx.doi.org/10.1016/j.cell.2012.05.014; PMID: 22726440
- Gomez L, Thibault H, Gharib A, Dumont JM, Vuagniaux G, Scalfaro P, et al. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol Heart Circ Physiol 2007; 293:H1654 - 61; http://dx.doi.org/10.1152/ajpheart.01378.2006; PMID: 17557911
- Santos JB, Schauwecker PE. Protection provided by cyclosporin A against excitotoxic neuronal death is genotype dependent. Epilepsia 2003; 44:995 - 1002; http://dx.doi.org/10.1046/j.1528-1157.2003.66302.x; PMID: 12887430
- Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010; 330:1247 - 51; http://dx.doi.org/10.1126/science.1189157; PMID: 21030605
- Leyva JA, Bianchet MA, Amzel LM. Understanding ATP synthesis: structure and mechanism of the F1-ATPase (Review). [Review] Mol Membr Biol 2003; 20:27 - 33; http://dx.doi.org/10.1080/0968768031000066532; PMID: 12745923
- Galluzzi L, Morselli E, Kepp O, Tajeddine N, Kroemer G. Targeting p53 to mitochondria for cancer therapy. Cell Cycle 2008; 7:1949 - 55; http://dx.doi.org/10.4161/cc.7.13.6222; PMID: 18642442
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9:47 - 59; http://dx.doi.org/10.1038/nrm2308; PMID: 18097445
- Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, et al. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 2000; 101:389 - 99; http://dx.doi.org/10.1016/S0092-8674(00)80849-1; PMID: 10830166
- Rodić N, Oka M, Hamazaki T, Murawski MR, Jorgensen M, Maatouk DM, et al. DNA methylation is required for silencing of ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells 2005; 23:1314 - 23; http://dx.doi.org/10.1634/stemcells.2005-0119; PMID: 16051982
- Pasti L, Pozzan T, Carmignoto G. Long-lasting changes of calcium oscillations in astrocytes. A new form of glutamate-mediated plasticity. J Biol Chem 1995; 270:15203 - 10; http://dx.doi.org/10.1074/jbc.270.25.15203; PMID: 7797504
- Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res 2010; 70:1793 - 803; http://dx.doi.org/10.1158/0008-5472.CAN-09-3112; PMID: 20145152
- De Giorgi F, Ahmed Z, Bastianutto C, Brini M, Jouaville LS, Marsault R, et al. Targeting GFP to organelles. Methods Cell Biol 1999; 58:75 - 85; http://dx.doi.org/10.1016/S0091-679X(08)61949-4; PMID: 9891375
- Pinton P, Ferrari D, Magalhães P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, et al. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 2000; 148:857 - 62; http://dx.doi.org/10.1083/jcb.148.5.857; PMID: 10704437
- Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14:1237 - 43; http://dx.doi.org/10.1038/sj.cdd.4402148; PMID: 17431418
- Hoffmann J, Vitale I, Buchmann B, Galluzzi L, Schwede W, Senovilla L, et al. Improved cellular pharmacokinetics and pharmacodynamics underlie the wide anticancer activity of sagopilone. Cancer Res 2008; 68:5301 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-08-0237; PMID: 18593931
- Boehrer S, Adès L, Braun T, Galluzzi L, Grosjean J, Fabre C, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood 2008; 111:2170 - 80; http://dx.doi.org/10.1182/blood-2007-07-100362; PMID: 17925489
- Arnoult D. Apoptosis-associated mitochondrial outer membrane permeabilization assays. Methods 2008; 44:229 - 34; http://dx.doi.org/10.1016/j.ymeth.2007.11.003; PMID: 18314053