
Abstract
Silent information regulator type-1 (SIRT1) is the best-studied member of the Sirtuin (Sir2) family of nicotinamide dinucleotide (NAD)-dependent class III histone deacetylases (HDACs), but has not yet been explored in cutaneous T-cell lymphoma (CTCL). We analyzed five CTCL cell lines and lesional tissues using flow cytometry, immunostaining, immunoblotting, cell death, viability, and apoptosis assays, small-molecule inhibitors, and shRNA knockdown. We found strong SIRT1 expression among CTCL lines relative to normal lymphocytes. CTCL cells in lesional tissues also expressed SIRT1 strongly. SIRT1 knockdown resulted in reduced cellular metabolism and proliferation, increased apoptosis, and PARP cleavage products. Tenovin-1, which reversibly inhibits class III HDACs (SIRT1 and SIRT2), reduced SIRT enzymatic activity and SIRT1 expression and led to increased apoptosis. These alterations were accompanied by increased forkhead box O3 (FoxO3) in several cell lines and increased nuclear p53, as well as acetylated p53 in wtp53 MyLa CTCL line. A combination of class I/II and class III HDACIs (vorinostat and tenovin-1) produced significantly greater growth inhibition, cell death via apoptosis, as well as superior p53 promoter upregulation in wtp53 MyLa cells as compared with either agent alone. This occurred in a partially p53-dependent manner, as these effects were blunted by p53 knockdown. Our results indicate that SIRT1 is strongly expressed in CTCL. Its inhibition results in reduced growth and increased apoptosis of CTCL cells. Furthermore, our findings suggest that some CTCL patients, such as those with wtp53, might benefit more from treatment with a combination of different classes of HDACIs than with a single agent.
Introduction
Epigenetic modification of histones by acetylation and deacetylation is regulated by histone acetyl transferases (HATs) and histone deacetylases (HDACs), which play key roles in transcriptional regulation of normal and malignant cells.Citation1-Citation5 Acetylation of histones promotes DNA accessibility for transcriptional activation, and thus can modulate gene transcription at multiple molecular targets.Citation3,Citation6,Citation7 Vorinostat (suberoylanilide hydroxamic acid) was the first HDAC inhibitor (HDACI) approved by the FDA for the treatment of refractory cutaneous T-cell lymphoma (CTCL).Citation8,Citation9 It inhibits the enzyme activity of class I (HDAC1, HDAC2, and HDAC3) and class II (HDAC6) HDACs.Citation10-Citation13 Romidepsin (depsipeptide, FK228) was the second FDA approved HDACI for CTCL therapy.Citation14-Citation17
CTCLs represent a complex array of disorders with various manifestations and clinical courses. Mycosis fungoides (MF) and Sézary syndrome (SS) are forms of CTCL characterized by the presence of clonally expanded, skin-homing helper-effector memory T cells (MF) and central memory T cells (SS) exhibiting abnormal apoptotic control mechanisms.Citation18 Defects in extrinsic death receptors, their ligands, and/or their inhibitors cause abnormalities leading to apoptotic resistance in CTCL.Citation19-Citation21 Normalization of these factors may sensitize CTCL cells to programmed cell death.Citation22-Citation24
Silent information regulator-1 (SIRT1) belongs to the unique class III HDACs involved in the gene silencing of yeast.Citation25 The class III HDACs enzymes are nicotinamide adenine dinucleotide (NAD+)-dependent ADP-ribosyltransferases (ARTs) that deacetylate lysine residues on various proteins, which regulate a variety of cellular and metabolic functions, such as gene silencing.Citation26,Citation27 They are phylogenetically conserved from yeast to humans. The first non-histone substrate recognized for the mammalian Sir2 ortholog, SIRT1, was the tumor suppressor protein p53. Other downstream targets for SIRT1 include FoxO factors, NFκB, cl AP2, Ku70, AR, PPARγ, p63, and p73.Citation28 SIRT1 is a soluble protein found mainly in the nucleus and nucleolus with residual cytoplasmic localizationCitation29 and has been implicated in cancers.Citation30 Although literature suggests that SIRT1 can act as either a tumor promoter or a tumor suppressor,Citation31 the knockdown of SIRT1 expression stops the growth of various tumor cell lines.Citation32,Citation33 We found that SIRT1 is strongly expressed in CTCL cells and lesional skin biopsies, and that its inhibition reduces CTCL growth and viability. In some CTCL lines, this effect is maximized when class I/II and class III HDACIs are used in combination.
Results
SIRT1 protein expression in CTCL
Using an array of CTCL lines (HH, Hut78, MyLa, SeAx, and SZ4) we found that CTCL cells express SIRT1 strongly, as shown by immunoblot in . Resting peripheral blood mononucleated cell (PBMC) lymphocytes lacked detectable SIRT1 expression, whereas A375 melanoma and acute T-cell leukemia cell line Jurkat cells showed expression similar to CTCL cells. The expression levels were confirmed using flow cytometry (). Immunohistochemical images of cytopreparations are shown in . Details of the staining are stated in “Materials and Methods”. SIRT1 immunoreactivity was found mostly in the cytoplasm with the exception of Jurkat and SZ4 cells, which demonstrated both cytoplasmic and nuclear immunopositivity. This is in line with a previous report suggesting that in cancer cells SIRT1 is localized mainly in the cytoplasm, in contrast to nuclear localization in normal cells.Citation29
Figure 1. SIRT1 protein is overexpressed in CTCL cells. (A) Immunoblot analysis for endogenous SIRT1 expression was performed using normal blood (resting PBMC), 5 CTCL cell lines (HH, Hut78, MyLa, SeAx, and SZ4), T-ALL Jurkat, and A375 melanoma (the latter from a separate blot and separated by a line). Numbers show the relative density of protein bands normalized to loading control GAPDH. Blots shown are representative of 3 independent experiments. (B) Intracellular SIRT1 was analyzed by flow cytometric analysis. Overlaid curves represent SIRT1 (black line) compared with isotype controls (filled gray). (C) Immunocytochemical staining shows SIRT1 immunoreactivity in CTCL cytopreparations, developed with DAB chromogen and methylene blue counterstain and photographed with Nikon Eclipse Ti microscope and a high-performance CCD camera using with an oil immersion lens at 60× magnification. (D) Immunohistochemical SIRT1 expression in lesional skin from CTCL patients showing clusters of stained tumor cells in epidermis and dermis (white arrow heads). Staining protocol was similar to , and images were photographed at 40× magnification using an oil immersion lens.
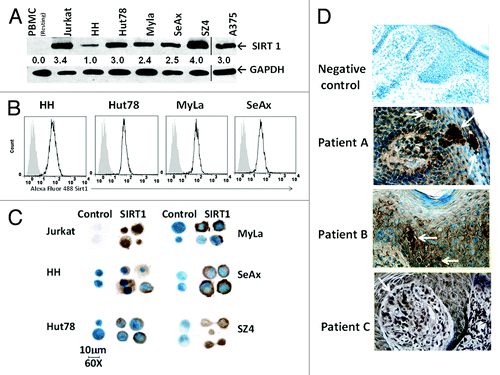
CTCL skin lesions also exhibited SIRT1 immunoreactivity as shown in . Fresh frozen biopsy samples (patients A–C) showed clusters of dark brown SIRT1+ tumor cells (with white arrows) when stained with DAB chromogen and methylene blue nuclear counterstain, along with negative control, as stated in “Materials and Methods”. Formalin-fixed and paraffin-embedded (FFPE) CTCL skin samples exhibited the same pattern (data not shown). Keratinocytes were also stained variably, serving as an internal positive control.
Effects of SIRT1 knockdown
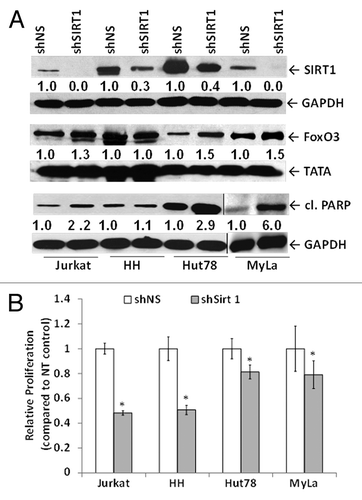
To understand the functional significance of SIRT1 in CTCL, we genetically knocked down SIRT1 using lentiviral shRNA interference. We used 4 cell lines, Jurkat, HH, Hut78, and MyLa, for most of these experiments. SIRT1 knockdown reduced the expression levels of SIRT1 in CTCL cells and Jurkat cells () at 48 h post-transduction. This reduction in SIRT1 expression was associated with increased FoxO3 levels and cleaved PARP products in Jurkat, Hut78, and MyLa cells (). The FoxO3 gene likely functions as a trigger for apoptosis through expression of genes necessary for cell death. SIRT1 knockdown also significantly reduced cellular metabolic activity at 48 h post-transduction, as determined by the MTT assay, thereby suggesting the role of SIRT1 in CTCL growth ().
Figure 2. SIRT1 knockdown in CTCL cells results in apoptosis and decreased cell proliferation associated with FoxO3 upregulation. (A) Lentiviral shRNA-mediated knockdown of SIRT1 results in reduced SIRT1 protein expression in whole-cell lysates of CTCL cells, upregulation of FoxO3 in nuclear lysates of Jurkat and CTCL Hut78 and MyLa cells, and PARP cleavage. The vertical line shows a separate blot. For densitometric analysis, the level of endogenous SIRT1, FoxO3, and cleaved PARP levels in shNS-transduced controls was considered as 1.0. Densitometric analysis of protein bands was performed relative to GAPDH loading control. (B) shRNA-mediated knockdown of SIRT1 reduces cellular metabolic activity. White bars represent shNS-transduced samples and gray bars represent shSIRT1-transduced samples. Data represented as mean proliferation ± standard deviation of 3 experiments with similar results (*P < 0.05).
Effects of SIRT1 small-molecule inhibitor, tenovin-1
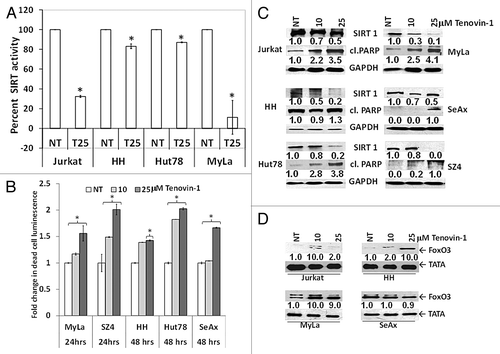
Chemical knockdown of SIRT1 was achieved using a benzoylthiourea compound, tenovin-1,Citation32 a reversible HDACI of SIRT1 and SIRT2 that functions upstream of p53 in a dose- and time-dependent manner. We assessed the inhibition of SIRT enzymatic activity at 24 h post-treatment with 25 μM tenovin-1. SIRT activity was reduced with tenovin-1 treatment, with greatest effect in Jurkat and MyLa cells (70 and 90% reduction, respectively), as compared with HH and Hut78 (10–20% reduction) (). Tenovin-1 treatment in a dose-dependent fashion resulted in reduced growth and viability of CTCL cells at 24–48 h, as a function of luminescence change, both in absolute numbers and normalized data as shown in . The viability-inhibitory effects of tenovin-1 were greatest at a 25-µM concentration after 24 h for MyLa and SZ4 (1.5- and 2-fold, respectively), and after 48 h for HH, Hut78, and SeAx (1.4-, 2-, and 1.7-fold, respectively). The individual growth kinetics of each CTCL line and the availability of drug at later time points probably accounts for differences in the extent of cell death between the 2 different time points. In we showed 2 time points together to show the best effects of the drug in different CTCL cells. Tenovin-1 treatment also caused a reduction in SIRT1 expression in a dose-dependent manner at 48 h that was associated with increased PARP cleavage in all CTCL cell lines and Jurkat, albeit to different extents (). Similar to the increased FoxO3 induced by SIRT1 knockdown (), tenovin-1 upregulated FoxO3 in Jurkat and some CTCL lines ().
Figure 3. Chemically induced SIRT1 inhibition causes cell death in association with reduced SIRT1 enzymatic activity, increased PARP cleavage, and increased FoxO3. (A) Tenovin-1 mediated reduction in SIRT enzymatic activity. Y-axis represents percent SIRT normalized luminescent activity compared with untreated control. (B) Tenovin-1 treatment reduces the viability of CTCL cells in a dose-dependent fashion that is maximal at 24 or 48 h depending on the specific CTCL line. White bars represent untreated controls and light and dark gray bars represent tenovin-1 at 10 and 25 μM doses, respectively. Data represented as mean ± standard deviation of 3 experiments with similar results (*P < 0.05). (C) Immunoblot analysis shows tenovin-1 mediated downregulation of SIRT1 expression, at 48 h post-treatment, is associated with PARP cleavage. (D) It is also associated with increased nuclear FoxO3. Numbers represent densitometric analysis of protein bands. For each cell line, the levels of endogenous SIRT1, cleaved PARP, and FoxO3 expression in untreated controls were considered as 1.0. Densitometric analysis of protein bands was performed relative to GAPDH/TATA loading control.
Effect of tenovin-1 on wild-type p53 in CTCL
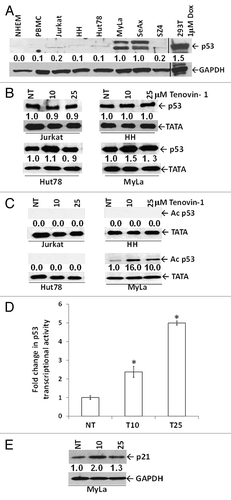
In addition to inhibition of histone deacetylation, HDACI are known to regulate a variety of other substrates, including p53. Among the CTCL lines used in this study, MyLa is the only one known to harbor wtp53. SeAx showed a mutation at codon 245, resulting in a Gly to Ser substitution.Citation34 Jurkat and Hut78 exhibited p53 mutations at codon 196, resulting in a stop signal and truncated protein.Citation34,Citation35 HH exhibited a mutated splice acceptor at exon 6, resulting in a stop at codon 190 and truncated protein.Citation34 All of these mutations were homozygous except for Jurkat, which was heterozygous. The p53 status of SZ4 cells has not been characterized. Using the antibody FL-393 that recognizes full-length p53, we detected a strong p53 band in immunoblots of MyLa and SeAx (). In contrast, antibody 7F5 2527, which recognizes the p53 N terminus, detected a p53 band in immunoblots of all cell lines (). Tenovin-1 treatment significantly upregulated total p53 expression levels only in MyLa in association with an increase in activated p53 (acetylated at Lys382) (). Next, we determined if the increase in total and acetylated p53 following tenovin-1 treatment was associated with activation of the p53 promoter. We found a statistically significant, 2–5-fold increase in promoter activity with 10 and 25 μM tenovin-1 (). Tenovin-1-mediated acetylation of p53 was also associated with an approximately 2-fold increase in downstream target p21 in MyLa with tenovin-1–10 μM, which, at 25-μM dose, was less pronounced due to the cytotoxic effects of drug ().
Figure 4. Tenovin-1 enhances p53 pathway signaling in wtp53 MyLa cells. (A) Immunoblot shows baseline expression levels of p53 using antibody (FL393) that detects full-length p53 of human origin. Densitometric analysis of protein bands was performed relative to GAPDH loading control. (B) Immunoblot using another antibody (7F5; 2527) specific for the N terminus of p53 shows that p53 is upregulated only in the cell line bearing wtp53 (MyLa). (C) Tenovin-1 treatment upregulates activated (acetylated) p53 expression only in wtp53 MyLa nuclear lysates. Densitometric analysis of protein bands was performed relative to TATA-binding protein (TATA) loading control. (D) Tenovin-1 treatment upregulates p53 promoter activity in wtp53 MyLa cells. Data represented as mean ± standard deviation of 3 experiments (6 replicates per experiment) with similar results (*P < 0.05). (E) Immunoblot shows that the upregulated p53 in MyLa cells is associated with upregulation of p21, a downstream target of p53. Densitometric analysis of protein bands was performed relative to GAPDH loading control.
Effect of combined HDAC class I, II, and III inhibition in CTCL
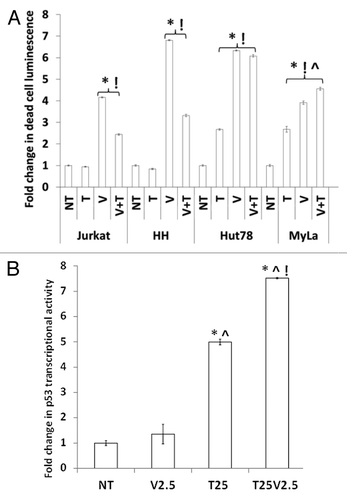
Because vorinostat inhibits both class I (HDAC1, 2, and 3) and II (HDAC6) HDACs, and tenovin-1 inhibits class III HDACs (SIRT1 and SIRT2), we used these 2 agents together to evaluate their combined effect on the viability of CTCL cells. This approach worked best for MyLa cells, which have an intact p53 function and showed a significantly higher cell death luminescence both in absolute numbers and in normalized data in response to the combination treatment as compared with monotherapy. In all other CTCL cells, 5 μM vorinostat was much more potent in causing cellular death than 25 μM tenovin-1 alone. The combination of vorinostat and tenovin-1 was not superior to vorinostat alone in Jurkat, HH, or Hut78 cells (). Using a luciferase reporter assay to assess p53 promoter activity in MyLa, we found a significant increase when vorinostat (2.5 μM) was combined with tenovin-1 at 25 μM compared with either agent alone (). This increase in p53 promoter activity parallels the increase in cell death observed with combination treatment of MyLa as shown in .
Figure 5. Effects of combined class I/II and class III HDACIs in CTCL. (A) Combination of different classes of HDACIs results in more cell death in wtp53 MyLa than either single agent. Statistical significance (P < 0.05) when compared with no treatment control (*), 25 μM tenovin-1, (!) and 5 μm vorinostat (^). (B) Increased transcriptional activity of p53 in the wtp53 MyLa CTCL line was greatest with combination of both classes of HDACIs. The histogram represents statistical significance when compared with no treatment control (*), 25 μM tenovin-1, (!) and 5 μm vorinostat (^) in 3 experiments with similar results (P < 0.05).
p53 knockdown blunts the effects of HDACI on wtp53 CTCL cells
To further understand the role of wtp53 in the response of MyLa to HDACI treatment, we knocked down p53 using shRNA interference. These altered MyLa cells () showed HDACI-mediated PARP cleavage, growth, and viability responses, at 48 h post-treatment, that were significantly blunted compared with control MyLa cells. For example, the significant 1.5-fold increase in dead cells induced by tenovin-1 at 24 h was abrogated by p53 knockdown to 1.1-fold ( vs. ). Furthermore, the ≥4-fold increases in dead cells induced by vorinostat and combination HDACI treatment were reduced to <2-fold ( vs. ). Similar reductions were noted in the generation of PARP cleavage products induced by tenovin-1 (; also compare with [4.1 vs. ~1]). These findings suggest that HDACI effects are at least partially p53-dependent in MyLa cells. Additionally, apoptotic measurement using annexin V binding assay also showed most cell death with the combination treatment ().
Figure 6. The p53 knockdown in MyLa cells blunts the effects of HDACIs. (A) MyLa cells (NT), with p53 knocked down by lentiviral control (shNS) and shp53. Tenovin-1, vorinostat, and combination-treated shNS and shp53 MyLa cells showed treatment-dependent apoptosis by cleaved PARP (89 kDa band). GAPDH served as loading control. (B) HDACI mediated increase in dead cell luminescence (gray bars) and decrease in live cell luminescence (white bars) compared with no treatment (NT) control (*), T25 (tenovin-1; 25 μM) (!), V5 (vorinostat; 5 μm) (^), VT (combination of V5 and T25). (C) shp53 MyLa cells treated as in 6B, stained with Annexin V and PI and analyzed by flow cytometry for apoptosis. (D) Immunoblot analysis shows the effect of single and combination treatments on protein levels of PUMA, p21, cleaved caspase-3 and cleaved caspase-9 and in shNS and shp53 MyLa cells. Densitometric analysis of protein bands was performed relative to GAPDH loading control.
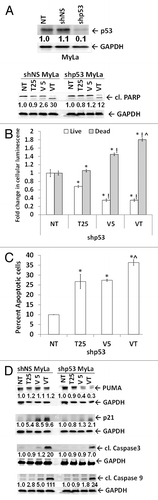
To determine if the extent of upregulation of p53 effectors by SIRT1 inhibition would be blunted when p53 was knocked down in MyLa cells, we looked at the expression levels of PUMA (p53 modulator of activity), p21, and cleaved caspases-3 and -9. The upregulation of these effectors by HDACIs was generally greatest when vorinostat and tenovin-1 were used in combination (). Furthermore, this upregulation was blunted by shp53 knockdown in MyLa cells.
Discussion
SIRT1 is implicated in a number of pathophysiological functions pertaining to aging, senescence, metabolic, and neurodegenerative disorders.Citation1,Citation27,Citation31 SIRT1 has a variety of substrates viz. NFκB, Ku70, p53, TFR2, PPARγ, p73, BcL-XL, MyoD, AR, cI AP2, and FoxO family of transcription factors.Citation1,Citation31 SIRT1 expression is a clinically significant, poor prognostic indicator for patients with diffuse large B-cell lymphoma.Citation36 In lymph nodes affected by classical Hodgkin lymphoma, the diagnostic Reed–Sternberg cells showed strong positivity for SIRT1.Citation36 SIRT1 is also highly expressed in acute adult T-cell leukemia (ATL) cells but not in chronic ATL. The SIRT1 inhibitor sirtinol induces ATL apoptosis in association with inhibition of NFκB signaling. These results suggest that SIRT1 is a crucial anti-apoptotic molecule in ATL cells and may be a significant marker for the conversion to acute ATL.Citation37
The role of SIRT1 in CTCL has not been explored previously. Therefore, in this study we evaluated its expression and function in this disorder. CTCL cells and lesional biopsies showed high SIRT1 expression and function. Reduced expression of SIRT1 in CTCL cells, achieved by shRNA knockdown, decreased CTCL metabolic activity, and increased apoptosis ( and ). Tenovin-1, a SIRT1 inhibitor, is a p53 activator that protects against MDM2-mediated p53 degradation, and thus elevates levels of p53 and p21 from an endogenous p53-dependent promoter. Tenovin-1 at 10 µM concentration upregulated p53 expression in MCF-7 cells within 2 h of treatment, and longer-term exposure significantly decreased the growth of BL2 Burkitt lymphoma and ARN8 melanoma cells, the latter derived from the A375 cell line containing a p53-reporter gene that is induced by tenovin-1.Citation32 In CTCL, tenovin-1 caused reduced SIRT1 expression and enzymatic activity, decreased cellular viability, and increased apoptosis, as evident by generation of PARP cleavage products ().
The protein level of p53 is regulated by SIRT1. Protein p53 is stabilized by acetylation of Lys382 residue,Citation38 which is a known target of SIRT1-mediated deacetylation.Citation1 As p53 plays a central role in critical cellular processes, including DNA repair, apoptosis, cell cycle arrest, senescence, and transformation, multiple pathways regulating cell fate may be effected by the action of class III HDACs on p53.Citation1 Although tenovin-1 increased levels of nuclear acetylated p53 only in wtp53 MyLa cells (), we found that growth-inhibiting and pro-apoptotic effects of tenovin-1 occurred in various CTCL cell lines lacking wtp53. Tenovin-1 treatment of MyLa cells upregulated p53 as well as acetylation of p53 at Lys382 in the nucleus, in accordance with previously published work on other cell types.Citation32 This upregulation and activation of p53 was found to be associated with transcriptional activation of the p53 promoter. When vorinostat (class I [HDAC1, 2, and 3] and II [HDAC6] HDACI) and tenovin-1 (class III [SIRT1 and SIRT2] HDACI) were combined to evaluate the total effect of inhibition of 3 different classes of HDACs, MyLa showed the greatest cell death response (). This prompted us to knockdown p53 in MyLa cells to evaluate the role of p53 status in this effect. p53 knockdown in MyLa cells exhibited less response than shNS controls, suggesting that the superior killing effects of the combination treatment were at least partially p53-dependent.
The FoxO3 gene belongs to the forkhead family of transcription factors, which are characterized by a distinct forkhead domain.Citation39 This gene may function as a trigger for apoptosis through expression of genes necessary for cell cycle arrest and cell death.Citation40 In the absence of p53 in MDA-MB-231 (mutant-p53) human breast cancer cell lines, FoxO3a was shown to be required for apoptotic activity.Citation41 Degradation of FoxO transcription factors is mediated by the ubiquitin proteasome pathway via phosphorylation and acetylation modifications.Citation40 Mammalian SIRT1 deacetylates FoxO3 and/or FoxO4, thus attenuating FoxO-induced apoptosis and potentiating survival.Citation39 This could explain the upregulation of FoxO3 in multiple CTCL lines when SIRT1 is inhibited by tenovin-1 or silenced via knockdown ( and ).
PUMA, a pro-apoptotic member of the BH3-only subgroup of the Bcl-2 family, is a key mediator of p53-dependent and p53-independent apoptosis.Citation42,Citation43 PUMA cooperates with direct activator proteins to induce mitochondrial outer membrane permeabilization and apoptosis via caspase activation.Citation44 Caspase-3 is either partially or totally responsible for the proteolytic cleavage of many key proteins such as PARP, and is therefore a critical executioner of apoptosis. As shown in , cleaved caspase-3 and -9 levels were also significantly upregulated by treatment. The p21 protein cleaves and activates caspases-6, -7, and -9, and the protein itself is processed by caspases-8, -9, and -10. Cleaved caspase-9 levels showed a gradual increase from tenovin-1 to vorinostat to combination treatment group ().
Class I/II HDACIs, such as romedepsin and vorinostat, are FDA-approved for the treatment of CTCL and have achieved overall response rates in the 30–40% range when used as monotherapy (8–16). Combination of these HDACIs with other agents, such as retinoids and interferons, has also proven beneficial.Citation45-Citation47 Our data suggest that enhanced tumor cell killing might be achievable by combining class I/II HDACIs with class III HDACIs, particularly in the treatment of tumors with intact wtp53. This type of combination therapy might also allow the use of individual agents at reduced doses, thereby minimizing side effects, such as immunosuppression observed with full doses of class I/II HDACIs.Citation46 Furthermore, we have shown recently that methotrexate (MTX) can be used clinically as a demethylating agent, resulting in the epigenetic de-repression of downregulated tumor suppressor genes such as FAS/CD95 in CTCL.Citation23,Citation48 Because DNA methylation and histone deacetylation are major epigenetic regulatory mechanisms for tumor suppressor gene silencing, the combination of MTX with HDACIs might constitute a novel therapeutic approach for CTCL.
It has been reported that tumor cells in most patients with leukemic CTCL (SS) have wtp53 but fail to initiate the downstream signal and induce p21.Citation34 The high level of SIRT1 in CTCL might prevent acetylation of p53 and thereby inhibit downstream signaling of the p53 cascade. If so, treatment with class III HDACIs might overcome this problem, resulting in enhanced tumor cell killing in SS patients. This view is supported by the positive effects of tenovin-1 on p53 downstream signaling observed in the wtp53 MyLa CTCL line and by the blunting of these effects following p53 knockdown.
Materials and Methods
Cell lines and treatments
Human CTCL lines MyLa, SeAx, and SZ4 were procured and grown in RPMI 1640 containing 2 mM L-glutamine, 1 mM pyruvate, and 10% fetal bovine serum as previously described.Citation23,Citation48 CTCL line HH and Hut78 and Jurkat are from the ATCC (CRL-2105, TIB-161, and TIB-152), and were grown in RPMI 1640 containing 2 mM L-glutamine, 1 mM pyruvate, 10% fetal bovine serum, and 10 mM HEPES. A375 melanoma and HEK294T cells were also from ATCC (CRL-1619 and CRL-11268) and grown in DMEM with 10% fetal bovine serum. Normal blood lymphocytes (PBMCs) were obtained from healthy donors as per institutional IRB protocol. Tenovin -1(N-[{(4-[acetylamino] phenyl) amino} thioxomethyl-4-{1,1-dimethylethyl}]-benzamide) was obtained from Cayman Chemical Company, dissolved in DMSO, and further diluted with the medium for the treatments at 10 and 25 μM doses. Vorinostat (Merck and Co) was dissolved in DMSO and used at 2.5- and 5-μM concentrations in corresponding media.
Immunoblot analysis
Following treatments, cells were washed with ice-cold PBS and lysed with RIPA buffer. For western blot analysis, 30 µg of protein samples were subjected to SDS-PAGE and transferred onto nitrocellulose membrane. Blots were exposed to SIRT1 and p53 (FL393 representing full-length) (Santa Cruz Biotechnology, Inc), FoxO3, PARP, p53 (7F5 representing NH2-terminus), Ac-p53 Lys 383, p21, PUMA, cleaved caspase-3, cleaved caspase-9, primary antibodies, and HRP conjugated appropriate secondary antibodies (Cell Signaling, Inc), followed by enhanced chemiluminescent detection (Thermo Fisher Scientific, Inc). Loading control antibodies were GAPDH and TATA binding protein (TBP) (Novus Biologicals and Abcam). The quantification of protein was performed by digital analysis of protein bands (TIFF images) using UN-SCAN-IT software (Silk Scientific, Inc). Densitometric analysis of protein bands was performed, and relative density of protein band segments was normalized to loading control GAPDH, and is expressed in numeric form below the immunoblots. Represented blots are from 3 independent experiments with similar results.
Flow cytometry
Control and treated cells were collected, washed with PBS, and fixed at 4 ° for 30 min with 2% paraformaldehyde. Thereafter cell pellets were resuspended in 0.5 ml of 0.1% saponin for permeabilization, washed, resuspended in 50 μl PBS containing SIRT1 antibody (1:50) for 60 min at room temperature, followed by washing and tagging with AlexaFluor 488 conjugated secondary antibody, and subjected to flow cytometry.
For Annexin V/PI-stained apoptosis analysis, control and treated cells were collected, washed with PBS, and then resuspended in 100 µl binding buffer (0.1 M Hepes, pH 7.4; 1.4 M NaCl; 25 mM CaCl2) and stained with FITC conjugated Annexin V antibody and propidium iodide (BD Biosciences) as per vendor’s suggestions. After staining, flow cytometric analysis was performed with a FACScan benchtop cytometer (BD Biosciences) at the UWCCC Flow Cytometry Facility using FlowJo software (Treestar).
Immunocytochemical/immunohistochemical staining
Immunocytochemical staining of the CTCL cells was performed on air-dried cell suspension droplets. Single-cell suspension of the cells was placed as a small drop on a glass slide (~8 mm diameter), air-dried, and stained as below, or stored in the freezer for later.
Immunohistochemical staining of CTCL lesions was performed using fresh frozen sections. Tissue sections were hydrated, blocked with endogenous peroxidase, incubated with SIRT1 monoclonal rabbit antibody (1:100) (Santa Cruz Biotechnology, Inc) at room temperature for an hour, washed with PBST after primary and secondary antibody incubations, using MACH 4 universal HRP-polymer detection system (Biocare Medical). Diaminobenzidine (DAB) was added for chromogen development and counterstained with methylene blue. Slides were gently wiped and mounted with 1:2 permount, dried, and photographed at 40× using a Nikon Eclipse Ti microscope (Nikon Instruments Inc). Cytoprepared cells on the glass slides were stained similarly and photographed at 60×.
Lentiviral production and transduction
HEK293T cells were passaged to be 30–40% confluent the following day. Cells were transfected with MISSION pLKO.1-puro (SHC001V) as a shNS-negative control or shSIRT1 (TRCN0000229680/ 81/ 83) or shp53 (TRCN000003753/ 56), as reported earlier.Citation49,Citation50 The efficacy of each batch of SIRT1 and p53 shRNA clones derived particles was about 70% and tested via immunoblot analysis for knockdown expression prior to further experiments.
MTT assay
For metabolic activity assessment of control and shSIRT1 transduced cells, cells were replated at the density of 1 × 103 in 100 μl volume in a 96-well plate (n = 3 or more). At 48 h post-plating, 20 μl of CellTiter Aqueous One solution reagent (Promega Corporation Inc) was added directly to the wells, incubated for 1 h at 37 °C in humidified 5% CO2, and absorbance was recording at 495 nm using a 96-well plate reader. The quantity of formazan product as measured by the amount of 495-nm absorbance is directly proportional to the number of living cells in culture, a marker for metabolic health of the cells. Data were represented as percent relative proliferation compared with shNS control as mean ± standard deviation of 3 experiments with similar results (*P < 0.05).
Cell growth and viability assay
Cellular growth and viability, after various treatments, was performed using CytoTox-Glo assay system (Promega Inc) as per manufacturer’s protocol. Briefly 10 × 103 cells in 100 μl volume in a 96-well plate were treated accordingly (n = 3–12). Post-treated cells were incubated with AAF-Glo substrate (alanyl-alanylphenylalanyl-aminoluciferin), which measures a distinct intracellular protease activity associated with cytotoxicity (dead-cell protease), for 15 min to measure “dead-cell” protease activity luminescent signal. With the addition of the lysis reagent, luminescent signal associated with the total number of cells in each assay well was measured 15 min later. Cell viability was calculated by subtracting the luminescent signal resulting from experimental cell death from total luminescent values. Data were represented as percent reduction in viable cells and percent increase in dead cells normalized to no treatment control.
SIRT activity
SIRT enzyme activity was measured using SIRT-Glow assay and screening system (Promega Inc). This assay is a single-reagent-addition, homogeneous, luminescent assay that measures the relative activity of the (NAD+)-dependent HDAC class III SIRTs. For assay, 20 × 103 cells/well were plated in 25 μl medium with or without tenovin-1 and incubated for 24 h. Subsequently 25 μl of assay reagent was added to each well, and the plates were incubated at room temperature for 15 min, in dark, followed by luminescent signal measurement. Data were represented as percent reduction in SIRT activity, normalized to no treatment control.
p53 luciferase promoter activity
Transcriptional activity of p53 was assayed using a Cignal reporter assay system (CCS-004L) as per SA Biosciences protocol. The dual-luciferase assay has a p53-specific firefly reporter and a Renilla luciferase normalization reporter. Briefly, 10 × 103 MyLa cells in 50 μl volume (n = 3–6), in 96-well plate, were transfected using Lipofectamine 2000 and p53 promoter DNA along with corresponding positive and negative control DNA in respective wells. After 24 h of transfection, cells were treated separately with 10 and 25 µM tenovin-1 or the combination with vorinostat, for 24 h and assayed as per Promega Dual Glow assay protocol according to the manufacturer’s protocol.
| Abbreviations: | ||
| ARTs | = | ADP-ribosyltransferases |
| CTCL | = | cutaneous T-cell lymphoma |
| DAB | = | diaminobenzidine |
| FoxO | = | forkhead box O |
| HATs | = | histone acetyl transferases |
| HDACs | = | histone deacetylases |
| HDACI | = | histone deacetylases inhibitor |
| MF | = | mycosis fungoides |
| NAD | = | nicotinamide dinucleotide |
| MTX | = | methotrexate |
| PBMC | = | peripheral blood mononucleated cell |
| PBS | = | phosphate buffer saline |
| SIRT1 | = | silent information regulator type-1 |
| SS | = | Sézary Syndrome |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Supported by Merit Review funding from the Department of Veterans Affairs (GSW and NA) and NCI (P30CA014520 and CA164417).
References
- Dai Y, Faller DV. Transcription Regulation by Class III Histone Deacetylases (HDACs)-Sirtuins. Transl Oncogenomics 2008; 3:53 - 65; PMID: 21566744
- Hagelkruys A, Sawicka A, Rennmayr M, Seiser C. The biology of HDAC in cancer: the nuclear and epigenetic components. Handb Exp Pharmacol 2011; 206:13 - 37; http://dx.doi.org/10.1007/978-3-642-21631-2_2; PMID: 21879444
- Johnsson AE, Wright AP. The role of specific HAT-HDAC interactions in transcriptional elongation. Cell Cycle 2010; 9:467 - 71; http://dx.doi.org/10.4161/cc.9.3.10543; PMID: 20081370
- Mehnert JM, Kelly WK. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J 2007; 13:23 - 9; http://dx.doi.org/10.1097/PPO.0b013e31803c72ba; PMID: 17464243
- Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene 2007; 26:6697 - 714; http://dx.doi.org/10.1038/sj.onc.1210755; PMID: 17934479
- Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol 2011; 2011:371832; http://dx.doi.org/10.1155/2011/371832; PMID: 21151613
- Schneider G, Krämer OH, Schmid RM, Saur D. Acetylation as a transcriptional control mechanism-HDACs and HATs in pancreatic ductal adenocarcinoma. J Gastrointest Cancer 2011; 42:85 - 92; http://dx.doi.org/10.1007/s12029-011-9257-1; PMID: 21271301
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109:31 - 9; http://dx.doi.org/10.1182/blood-2006-06-025999; PMID: 16960145
- Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs 2007; 16:1111 - 20; http://dx.doi.org/10.1517/13543784.16.7.1111; PMID: 17594194
- Duvic M, Olsen EA, Breneman D, Pacheco TR, Parker S, Vonderheid EC, Abuav R, Ricker JL, Rizvi S, Chen C, et al. Evaluation of the long-term tolerability and clinical benefit of vorinostat in patients with advanced cutaneous T-cell lymphoma. Clin Lymphoma Myeloma 2009; 9:412 - 6; http://dx.doi.org/10.3816/CLM.2009.n.082; PMID: 19951879
- Rangwala S, Zhang C, Duvic M. HDAC inhibitors for the treatment of cutaneous T-cell lymphomas. Future Med Chem 2012; 4:471 - 86; http://dx.doi.org/10.4155/fmc.12.6; PMID: 22416775
- Sato A. Vorinostat approved in Japan for treatment of cutaneous T-cell lymphomas: status and prospects. Onco Targets Ther 2012; 5:67 - 76; http://dx.doi.org/10.2147/OTT.S23874; PMID: 22573938
- Zhang C, Richon V, Ni X, Talpur R, Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol 2005; 125:1045 - 52; http://dx.doi.org/10.1111/j.0022-202X.2005.23925.x; PMID: 16297208
- Frye R, Myers M, Axelrod KC, Ness EA, Piekarz RL, Bates SE, Booher S. Romidepsin: a new drug for the treatment of cutaneous T-cell lymphoma. Clin J Oncol Nurs 2012; 16:195 - 204; http://dx.doi.org/10.1188/12.CJON.195-204; PMID: 22459529
- Haigentz M Jr., Kim M, Sarta C, Lin J, Keresztes RS, Culliney B, Gaba AG, Smith RV, Shapiro GI, Chirieac LR, et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol 2012; 48:1281 - 8; http://dx.doi.org/10.1016/j.oraloncology.2012.05.024; PMID: 22748449
- Kim M, Thompson LA, Wenger SD, O’Bryant CL. Romidepsin: a histone deacetylase inhibitor for refractory cutaneous T-cell lymphoma. Ann Pharmacother 2012; 46:1340 - 8; http://dx.doi.org/10.1345/aph.1R036; PMID: 22968522
- Prince HM, Dickinson M. Romidepsin for cutaneous T-cell lymphoma. Clin Cancer Res 2012; 18:3509 - 15; http://dx.doi.org/10.1158/1078-0432.CCR-11-3144; PMID: 22535155
- Stutz N, Johnson RD, Wood GS. The Fas apoptotic pathway in cutaneous T-cell lymphomas: frequent expression of phenotypes associated with resistance to apoptosis. J Am Acad Dermatol 2012; 67:e1 - 10; PMID: 22884443
- Nevala H, Karenko L, Vakeva L, Ranki A. Proapoptotic and antiapoptotic markers in cutaneous T-cell lymphoma skin infiltrates and lymphomatoid papulosis. Br J Dermatol 2001; 145:928 - 37; http://dx.doi.org/10.1046/j.1365-2133.2001.04523.x; PMID: 11899146
- Ni X, Zhang C, Talpur R, Duvic M. Resistance to activation-induced cell death and bystander cytotoxicity via the Fas/Fas ligand pathway are implicated in the pathogenesis of cutaneous T cell lymphomas. J Invest Dermatol 2005; 124:741 - 50; http://dx.doi.org/10.1111/j.0022-202X.2005.23657.x; PMID: 15816832
- Vermeer MH, van Doorn R, Dukers D, Bekkenk MW, Meijer CJ, Willemze R. CD8+ T cells in cutaneous T-cell lymphoma: expression of cytotoxic proteins, Fas Ligand, and killing inhibitory receptors and their relationship with clinical behavior. J Clin Oncol 2001; 19:4322 - 9; PMID: 11731515
- Contassot E, French LE. Targeting apoptosis defects in cutaneous T-cell lymphoma. J Invest Dermatol 2009; 129:1059 - 61; http://dx.doi.org/10.1038/jid.2009.14; PMID: 19369931
- Wu J, Wood GS. Reduction of Fas/CD95 promoter methylation, upregulation of Fas protein, and enhancement of sensitivity to apoptosis in cutaneous T-cell lymphoma. Arch Dermatol 2011; 147:443 - 9; http://dx.doi.org/10.1001/archdermatol.2010.376; PMID: 21173302
- Wu J, Nihal M, Siddiqui J, Vonderheid EC, Wood GS. Low FAS/CD95 expression by CTCL correlates with reduced sensitivity to apoptosis that can be restored by FAS upregulation. J Invest Dermatol 2009; 129:1165 - 73; http://dx.doi.org/10.1038/jid.2008.309; PMID: 18923451
- Carafa V, Nebbioso A, Altucci L. Sirtuins and disease: the road ahead. Front Pharmacol 2012; 3:4; http://dx.doi.org/10.3389/fphar.2012.00004; PMID: 22319497
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 2012; 13:225 - 38; PMID: 22395773
- Zschoernig B, Mahlknecht U. SIRTUIN 1: regulating the regulator. Biochem Biophys Res Commun 2008; 376:251 - 5; http://dx.doi.org/10.1016/j.bbrc.2008.08.137; PMID: 18774777
- Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 2007; 26:5489 - 504; http://dx.doi.org/10.1038/sj.onc.1210616; PMID: 17694089
- Byles V, Chmilewski LK, Wang J, Zhu L, Forman LW, Faller DV, Dai Y. Aberrant cytoplasm localization and protein stability of SIRT1 is regulated by PI3K/IGF-1R signaling in human cancer cells. Int J Biol Sci 2010; 6:599 - 612; http://dx.doi.org/10.7150/ijbs.6.599; PMID: 20941378
- Bosch-Presegué L, Vaquero A. The dual role of sirtuins in cancer. Genes Cancer 2011; 2:648 - 62; http://dx.doi.org/10.1177/1947601911417862; PMID: 21941620
- Deng CX. SIRT1, is it a tumor promoter or tumor suppressor?. Int J Biol Sci 2009; 5:147 - 52; http://dx.doi.org/10.7150/ijbs.5.147; PMID: 19173036
- Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008; 13:454 - 63; http://dx.doi.org/10.1016/j.ccr.2008.03.004; PMID: 18455128
- Olmos Y, Brosens JJ, Lam EW. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updat 2011; 14:35 - 44; http://dx.doi.org/10.1016/j.drup.2010.12.001; PMID: 21195657
- Lamprecht B, Kreher S, Möbs M, Sterry W, Dörken B, Janz M, Assaf C, Mathas S. The tumour suppressor p53 is frequently nonfunctional in Sézary syndrome. Br J Dermatol 2012; 167:240 - 6; http://dx.doi.org/10.1111/j.1365-2133.2012.10918.x; PMID: 22384858
- Cheng J, Haas M. Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol Cell Biol 1990; 10:5502 - 9; PMID: 2144611
- Jang KY, Hwang SH, Kwon KS, Kim KR, Choi HN, Lee NR, Kwak JY, Park BH, Park HS, Chung MJ, et al. SIRT1 expression is associated with poor prognosis of diffuse large B-cell lymphoma. Am J Surg Pathol 2008; 32:1523 - 31; http://dx.doi.org/10.1097/PAS.0b013e31816b6478; PMID: 18724249
- Kozako T, Aikawa A, Shoji T, Fujimoto T, Yoshimitsu M, Shirasawa S, Tanaka H, Honda S, Shimeno H, Arima N, et al. High expression of the longevity gene product SIRT1 and apoptosis induction by sirtinol in adult T-cell leukemia cells. Int J Cancer 2012; 131:2044 - 55; http://dx.doi.org/10.1002/ijc.27481; PMID: 22322739
- Lane DP, Cheok CF, Lain S. p53-based cancer therapy. Cold Spring Harb Perspect Biol 2010; 2:a001222; http://dx.doi.org/10.1101/cshperspect.a001222; PMID: 20463003
- Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol 2004; 14:408 - 12; http://dx.doi.org/10.1016/j.tcb.2004.07.006; PMID: 15308206
- Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci 2007; 120:2479 - 87; http://dx.doi.org/10.1242/jcs.001222; PMID: 17646672
- Lam M, Carmichael AR, Griffiths HR. An aqueous extract of Fagonia cretica induces DNA damage, cell cycle arrest and apoptosis in breast cancer cells via FOXO3a and p53 expression. PLoS One 2012; 7:e40152; http://dx.doi.org/10.1371/journal.pone.0040152; PMID: 22761954
- Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene 2008; 27:Suppl 1 S71 - 83; http://dx.doi.org/10.1038/onc.2009.45; PMID: 19641508
- Hikisz P, Kiliańska ZM. PUMA, a critical mediator of cell death-one decade on from its discovery. Cell Mol Biol Lett 2012; 17:646 - 69; http://dx.doi.org/10.2478/s11658-012-0032-5; PMID: 23001513
- Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochem Soc Trans 2001; 29:684 - 8; http://dx.doi.org/10.1042/BST0290684; PMID: 11709054
- Nolan L, Johnson PW, Ganesan A, Packham G, Crabb SJ. Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential?. Br J Cancer 2008; 99:689 - 94; http://dx.doi.org/10.1038/sj.bjc.6604557; PMID: 18728657
- Samimi S, Morrissey K, Anshelevich S, Evans K, Gardner J, Musiek A, Vittorio C, Rook A, Kim E. Romidepsin and interferon gamma: a novel combination for refractory cutaneous T-cell lymphoma. J Am Acad Dermatol 2013; 68:e5 - 6; http://dx.doi.org/10.1016/j.jaad.2011.06.043; PMID: 23244387
- Zhang X, Han S, Kang Y, Guo M, Hong S, Liu F, Fu S, Wang L, Wang QX. SAHA, an HDAC inhibitor, synergizes with tacrolimus to prevent murine cardiac allograft rejection. Cell Mol Immunol 2012; 9:390 - 8; http://dx.doi.org/10.1038/cmi.2012.28; PMID: 22922441
- Wu J, Siddiqui J, Nihal M, Vonderheid EC, Wood GS. Structural alterations of the FAS gene in cutaneous T-cell lymphoma (CTCL). Arch Biochem Biophys 2011; 508:185 - 91; http://dx.doi.org/10.1016/j.abb.2010.10.020; PMID: 21036138
- Nihal M, Stutz N, Schmit T, Ahmad N, Wood GS. Polo-like kinase 1 (Plk1) is expressed by cutaneous T-cell lymphomas (CTCLs), and its downregulation promotes cell cycle arrest and apoptosis. Cell Cycle 2011; 10:1303 - 11; http://dx.doi.org/10.4161/cc.10.8.15353; PMID: 21436619
- Schmit TL, Nihal M, Ndiaye M, Setaluri V, Spiegelman VS, Ahmad N. Numb regulates stability and localization of the mitotic kinase PLK1 and is required for transit through mitosis. Cancer Res 2012; 72:3864 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-12-0714; PMID: 22593191