
Abstract
Despite intense studies, highly effective therapeutic strategies against cancer have not yet been fully exploited, because few true cancer-specific targets have been identified. Most modalities, perhaps with the exception of radiation therapy, target proliferating cells, which are also abundant in normal tissues. Thus, most current cancer treatments have significant side effects. More than 10 years ago, the tumor suppressor p53 was first explored as a cancer-specific target. At the time, the approach was to introduce a normal p53 gene into mutant p53 (mp53) tumor cells to induce cell cycle arrest and apoptosis. However, this strategy did not hold up and mostly failed in subsequent clinical studies. Recent research developments have now returned p53 to the limelight. Several studies have reported that mutant or null p53 tumor cells undergo apoptosis more easily than genetically matched, normal p53 counterparts when inhibiting a specific stress kinase in combination with standard chemotherapy or when exposed to an ataxia-telangiectasia mutated (ATM) kinase inhibitor and radiation, thus achieving true cancer specificity in animal tumor models. This short review highlights several of these recent studies, discusses possible mechanism(s) for mp53-mediated “synthetic lethality,” and the implications for cancer therapy.
p53-Based Cancer Therapy
Cancer is an extremely multi-faceted disease able to escape normal growth constraints through a stepwise mutagenic mechanism that, in most cases, results in refractory responses to chemo- and radiation therapies.Citation1 Tumor cells adapt to unperturbed growth and survival by oncogene and non-oncogene “addiction”, making them highly dynamic and evasive in their responses to treatment.Citation2,Citation3 Recent work has demonstrated that, despite this adaptability, tumor cells have unique cancer signatures which provide an “Achilles’ heel” suitable for therapeutic targeting. The concept of “synthetic lethality”Citation4 was first realized in human cells when it was discovered that breast and ovarian tumor cells with BRCA1 and BRCA2 mutations—rendering them defective in homologous recombination—were sensitive to PARP inhibitors.Citation5,Citation6 However, BRCA mutations are relatively rare, so other, more frequently occurring cancer-specific targets need to be identified and exploited for therapeutic intervention.
About 50% of all human cancers have mutations in the p53 tumor suppressor gene.Citation7 In the late 1990s and early 2000s, a therapeutic strategy was developed based on the idea that introducing a normal p53 gene into mp53 tumor cells would restore their ability to undergo cell cycle arrest, apoptosis, and differentiation in response to chemo- and radiotherapy. However, inadequate gene delivery and the continued presence of mp53 in the tumor cells promoting genomic instability led to a mutation-prone phenotype and subsequent escape from the effects of the exogenously introduced normal p53. These factors likely contributed to the failure of p53 gene therapy, even though such efforts continue to this day.Citation8-Citation10 Currently, the focus in the field is to restore normal p53 function of the endogenous mutant form with small molecules.Citation11
Mutant p53-Based Therapeutic Strategies
Recently, several groups have reported on the increased sensitivity of mp53 tumor cells to chemo- and radiation therapy. In a series of elegant studies, it was demonstrated that the p38 MAP kinase-MAPKAP kinase-2 (MK2) signaling node complements the well-established ATM-Chk2 and ATR-Chk1 nodes converging on the cell cycle regulator Cdc25 during the DNA damage response (DDR).Citation12,Citation13 The mechanism by which p38-MK2 regulates the DDR in an mp53-dependent manner was shown to go through the G1/S and G2/M checkpoints, resulting in synthetic lethality.Citation14 Downregulation of MK2 in p53-null cells, but not in cells with normal p53, resulted in mitotic catastrophe and apoptosis after treatment with chemotherapy.Citation12 In an innovative study, Morandell et al. generated conditional knockout mice in which both MK2+ and MK2− tumors could be generated within the same animal. Combining this technique with an autochthonous non-small cell lung cancer model, the investigators were able to establish that knocking out MK2 in p53-null tumors resulted in a much improved in vivo response to cisplatin exposure when compared with the same treatment of tumors with normal p53.Citation13 Thus, a strategy to specifically enhance apoptosis of p53-deficient tumors by targeting MK2 was identified. Small-molecule p38 MAP kinase and/or MK2 inhibitors developed primarily for controlling inflammation and other stress conditions might therefore benefit cancer patients with p53-deficient or mutant tumors undergoing standard chemotherapy.Citation13
Whereas the work discussed above was done with chemotherapeutics, our group recently examined the response of glioma cells and tumors to radiosensitization using an ATM kinase inhibitor.Citation15 Similar to the MK2 studies, glioma cells and tumors expressing mp53 were much more responsive to radiosensitization than isogenic wild-type p53 cells and tumors. Using genetically matched, orthotopically implanted glioma cells, we were able to demonstrate significantly increased in vivo survival after concurrent treatment with ATM kinase inhibition and radiation in the mp53 vs. the wild-type group. Impressively, the dual-treatment mp53 cohort survived, on average, for several months longer than their wild-type counterparts. While there is a clear advantage to using ATM inhibition concomitant with standard chemoradiation for gliomas, it is, at this point, unclear as to whether this differential p53 response is also dependent on MK2. If so, ATM kinase inhibition could potentially block TAO-p38-MK2 signaling and inhibit MK2 activation, since ATM and ATR directly phosphorylate the TAO kinases in response to DNA damage.Citation16 Activated TAO kinases, in turn, phosphorylate p38 in a complex with MK2, resulting in the release and activation of MK2. Possible mechanisms for the increased response of mp53 tumor cells to chemo- and radiation therapy are outlined in . It is currently unclear whether this differential p53 response to ATMi radiosensitzation is unique to gliomas or is of a more general nature. It is, however, known that replicative stress is elevated in gliomas, contributing to aberrant constitutive activation of DNA damage signaling and perhaps sensitizing them to such treatment.Citation17 Another study, using an ATM kinase inhibitor derived from the same first-generation analog as ours,Citation18 described no such p53 dependence on a variety of other types of tumor cells.Citation19 However, a side-by-side in vivo comparison using matched p53 tumors was not done in this study. Regardless of whether or not ATM inhibitor radiosensitization occurs by an MK2-dependent mechanism, if patients with mp53 gliomas are as responsive as tumors in pre-clinical models,Citation15 a critical, cancer-specific effect might be achieved. This mp53-specific effect could open up an enhanced therapeutic window, which would enable a reduction in overall radiation dose and sparing of normal brain. Of course, mp53-targeted strategies could perhaps be further enhanced with “cyclotherapy” or “time-staggered” drug approaches.Citation20-Citation23
Figure 1. Possible mechanism(s) of ATM inhibitor-mediated mp53 radiosensitization. See text for explanation. For simplicity, focus is on ATM signaling nodes. Abbreviations: ATMi, ATM kinase inhibitor; IR, ionizing radiation; MK2i, MK2 inhibitor; p38i, p38 MAP kinase inhibitor. Arrows denote activation and “T”s dephosphorylation/inhibition. Shapes outlined in red denote phosphorylation events. Adapted from references Citation14 and Citation16.
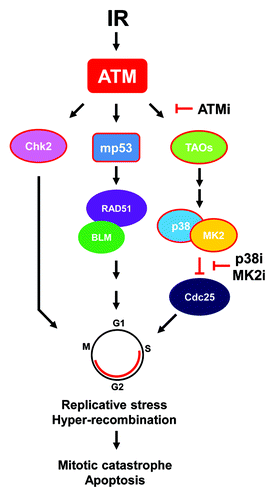
It has long been known that p53 suppresses homologous recombination by direct binding to RAD51 and the BLM helicase that protects against replication-induced DNA double-strand breaks.Citation24-Citation26 Conversely, mp53 promotes basal and DNA damage-induced increases in RAD51 that results in “hyper-recombination”.Citation27,Citation28 Thus, the heightened sensitivity to an ATM kinase inhibitor and increased radiosensitivity of mp53 glioma cells could be the result of aberrant recombination occurring in these cells or, alternatively, that cells with normal p53 are protected (or both). A few years back, we observed that a p38 MAP kinase inhibitor stimulated homologous recombination,Citation29 suggesting that blocking p38 might result in hyper-recombination and, consequently, links the inhibition of p38-MK2 to aberrant DNA repair (). In support of this finding, it was recently shown that DNA damage-induced replication fork stalling is dependent on MK2,Citation30 a finding which might link the mp53 abrogation of S-phase checkpoint control with trans-lesion replication and hyper-recombination when MK2 activity is blocked. It is worth noting that the role of p53 in replication fork stalling was not determined in this study. Interestingly, we reported several years ago that BRCA1 mutants unable to bind A, B, and C complexes through the BRCT domain demonstrated a hyper-recombination phenotype.Citation31,Citation32 Since BRCA1 and p53 are intimately linked by direct interaction via the N-terminal BARD and C-terminal BRCT domains, and mp53 is unable to support BRCA1 nuclear export,Citation33,Citation34 it is possible that mp53-directed hyper-recombination is related to mutant BRCA1 hyper-recombination. Altogether, whereas the consequence of MK2- or ATM-targeted therapies of mp53 tumors is relatively clear, i.e., apoptosis through checkpoint failure, the underlying mechanism is not.
Conclusions
Regardless of the mechanism behind the increased response of mp53 tumors to chemo- and radiation therapy by MK2 knockdown(out) and small-molecule ATM kinase inhibitors, it is likely that these findings will spur a closer look at stratifying patients according to p53 status in future clinical trials. In theory, half of all cancers (those with mp53) might be candidates for adjuvant therapy targeting MK2 and ATM. An advantage with the latter is that a single agent would not only provide highly effective radiosensitization (ideally through conformal radiation therapy), but might also inhibit tumor dispersal as well as specifically target cancer.Citation15,Citation35,Citation36
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Supported in part by NIH R01NS064593 and R21CA156995 (K.V.), and F30CA171893 (J.M.B.).
References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70; http://dx.doi.org/10.1016/S0092-8674(00)81683-9; PMID: 10647931
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science 2002; 297:63 - 4; http://dx.doi.org/10.1126/science.1073096; PMID: 12098689
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009; 136:823 - 37; http://dx.doi.org/10.1016/j.cell.2009.02.024; PMID: 19269363
- Kaelin WG Jr.. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5:689 - 98; http://dx.doi.org/10.1038/nrc1691; PMID: 16110319
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913 - 7; http://dx.doi.org/10.1038/nature03443; PMID: 15829966
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917 - 21; http://dx.doi.org/10.1038/nature03445; PMID: 15829967
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253:49 - 53; http://dx.doi.org/10.1126/science.1905840; PMID: 1905840
- Roth JA. Adenovirus p53 gene therapy. Expert Opin Biol Ther 2006; 6:55 - 61; http://dx.doi.org/10.1517/14712598.6.1.55; PMID: 16370914
- Swisher SG, Roth JA, Komaki R, Gu J, Lee JJ, Hicks M, Ro JY, Hong WK, Merritt JA, Ahrar K, et al. Induction of p53-regulated genes and tumor regression in lung cancer patients after intratumoral delivery of adenoviral p53 (INGN 201) and radiation therapy. Clin Cancer Res 2003; 9:93 - 101; PMID: 12538456
- Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer?. Lancet Oncol 2003; 4:415 - 22; http://dx.doi.org/10.1016/S1470-2045(03)01139-2; PMID: 12850192
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009; 9:862 - 73; http://dx.doi.org/10.1038/nrc2763; PMID: 19935675
- Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007; 11:175 - 89; http://dx.doi.org/10.1016/j.ccr.2006.11.024; PMID: 17292828
- Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, Couvillon AD, Jacks T, Yaffe MB. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep 2013; 5:868 - 77; http://dx.doi.org/10.1016/j.celrep.2013.10.025; PMID: 24239348
- Reinhardt HC, Jiang H, Hemann MT, Yaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle 2009; 8:3112 - 9; http://dx.doi.org/10.4161/cc.8.19.9626; PMID: 19755856
- Biddlestone-Thorpe L, Sajjad M, Rosenberg E, Beckta JM, Valerie NC, Tokarz M, Adams BR, Wagner AF, Khalil A, Gilfor D, et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin Cancer Res 2013; 19:3189 - 200; http://dx.doi.org/10.1158/1078-0432.CCR-12-3408; PMID: 23620409
- Raman M, Earnest S, Zhang K, Zhao Y, Cobb MH. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J 2007; 26:2005 - 14; http://dx.doi.org/10.1038/sj.emboj.7601668; PMID: 17396146
- Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010; 29:5095 - 102; http://dx.doi.org/10.1038/onc.2010.249; PMID: 20581868
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-04-2727; PMID: 15604286
- Batey MA, Zhao Y, Kyle S, Richardson C, Slade A, Martin NM, Lau A, Newell DR, Curtin NJ. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol Cancer Ther 2013; 12:959 - 67; http://dx.doi.org/10.1158/1535-7163.MCT-12-0707; PMID: 23512991
- Blagosklonny MV. How cancer could be cured by 2015. Cell Cycle 2005; 4:269 - 78; http://dx.doi.org/10.4161/cc.4.2.1493; PMID: 15655345
- van Leeuwen IM. Cyclotherapy: opening a therapeutic window in cancer treatment. Oncotarget 2012; 3:596 - 600; PMID: 22711025
- Rao B, Lain S, Thompson AM. p53-Based cyclotherapy: exploiting the ‘guardian of the genome’ to protect normal cells from cytotoxic therapy. Br J Cancer 2013; 109:2954 - 8; http://dx.doi.org/10.1038/bjc.2013.702; PMID: 24231949
- Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012; 149:780 - 94; http://dx.doi.org/10.1016/j.cell.2012.03.031; PMID: 22579283
- Stürzbecher HW, Donzelmann B, Henning W, Knippschild U, Buchhop S. p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction. EMBO J 1996; 15:1992 - 2002; PMID: 8617246
- Sengupta S, Linke SP, Pedeux R, Yang Q, Farnsworth J, Garfield SH, Valerie K, Shay JW, Ellis NA, Wasylyk B, et al. BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J 2003; 22:1210 - 22; http://dx.doi.org/10.1093/emboj/cdg114; PMID: 12606585
- Kumari A, Schultz N, Helleday T. p53 protects from replication-associated DNA double-strand breaks in mammalian cells. Oncogene 2004; 23:2324 - 9; http://dx.doi.org/10.1038/sj.onc.1207379; PMID: 14743204
- Saintigny Y, Rouillard D, Chaput B, Soussi T, Lopez BS. Mutant p53 proteins stimulate spontaneous and radiation-induced intrachromosomal homologous recombination independently of the alteration of the transactivation activity and of the G1 checkpoint. Oncogene 1999; 18:3553 - 63; http://dx.doi.org/10.1038/sj.onc.1202941; PMID: 10380877
- Bertrand P, Rouillard D, Boulet A, Levalois C, Soussi T, Lopez BS. Increase of spontaneous intrachromosomal homologous recombination in mammalian cells expressing a mutant p53 protein. Oncogene 1997; 14:1117 - 22; http://dx.doi.org/10.1038/sj.onc.1200931; PMID: 9070661
- Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res 2007; 67:1046 - 53; http://dx.doi.org/10.1158/0008-5472.CAN-06-2371; PMID: 17283137
- Köpper F, Bierwirth C, Schön M, Kunze M, Elvers I, Kranz D, Saini P, Menon MB, Walter D, Sørensen CS, et al. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc Natl Acad Sci U S A 2013; 110:16856 - 61; http://dx.doi.org/10.1073/pnas.1304355110; PMID: 24082115
- Dever SM, White ER, Hartman MC, Valerie K. BRCA1-directed, enhanced and aberrant homologous recombination: mechanism and potential treatment strategies. Cell Cycle 2012; 11:687 - 94; http://dx.doi.org/10.4161/cc.11.4.19212; PMID: 22306997
- Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, Valerie N, Xu B, Povirk LF, Valerie K. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging (Albany NY) 2011; 3:515 - 32; PMID: 21666281
- Feng Z, Kachnic L, Zhang J, Powell SN, Xia F. DNA damage induces p53-dependent BRCA1 nuclear export. J Biol Chem 2004; 279:28574 - 84; http://dx.doi.org/10.1074/jbc.M404137200; PMID: 15087457
- Jiang J, Yang ES, Jiang G, Nowsheen S, Wang H, Wang T, Wang Y, Billheimer D, Chakravarthy AB, Brown M, et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Res 2011; 71:5546 - 57; http://dx.doi.org/10.1158/0008-5472.CAN-10-3423; PMID: 21742769
- Golding SE, Rosenberg E, Adams BR, Wignarajah S, Beckta JM, O’Connor MJ, Valerie K. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle 2012; 11:1167 - 73; http://dx.doi.org/10.4161/cc.11.6.19576; PMID: 22370485
- Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, Chong WY, Hummersone M, Rigoreau L, Menear KA, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther 2009; 8:2894 - 902; http://dx.doi.org/10.1158/1535-7163.MCT-09-0519; PMID: 19808981