
Abstract
Mutations of tumor suppressor Nf1 gene deregulate Ras-mediated signaling, which confers the predisposition for developing benign or malignant tumors. Inhibition of protein kinase C (PKC) was shown to be in synergy with aberrant Ras for the induction of apoptosis in various types of cancer cells. However, it has not been investigated whether loss of PKC is lethal for Nf1-deficient cells. In this study, using HMG (3-hydroxy-3-methylgutaryl, a PKC inhibitor), we demonstrate that the inhibition of PKC by HMG treatment triggered a persistently mitotic arrest, resulting in the occurrence of mitotic catastrophe in Nf1-deficient ST8814 cells. However, the introduction of the Nf1 effective domain gene into ST8814 cells abolished this mitotic crisis. In addition, HMG injection significantly attenuated the growth of the xenografted ST8814 tumors. Moreover, Chk1 was phosphorylated, accompanied with the persistent increase of cyclin B1 expression in HMG-treated ST8814 cells. The knockdown of Chk1 by the siRNA prevented the Nf1-deficient cells from undergoing HMG-mediated mitotic arrest as well as mitotic catastrophe. Thus, our data suggested that the suppression of PKC activates the Chk1-mediated mitotic exit checkpoint in Nf1-deficient cells, leading to the induction of apoptosis via mitotic catastrophe. Collectively, the study indicates that targeting PKC may be a potential option for developing new strategies to treat Nf1-deficiency-related diseases.
Introduction
Neurofibromin (the protein product of Nf1) shares the sequence similarities with the catalytic domain of p120 GAP.Citation1 Biochemical analysis of the recombinant peptides corresponding to this domain and crystallography revealed that Nf1 is a GAP (GTPase-activating proteins) for Ras.Citation1-Citation5 The inactivation of neurofibromin was shown to link to an increase in Ras activity in murine cells with Nf1 defect and some human Nf1 deficient tumor cells.Citation6-Citation8 The experimental information suggested that the growth advantage in these tumor cells was most likely conferred by hyperactivity of Ras signaling due to the loss of the function of neurofibromin.Citation1,Citation9-Citation12
The GAP activity of neurofibromin has profound implications in the pathology and complications of Neurofibromatosis type 1 (NF1) patients. This common familial tumor predisposition syndrome is inherited in an autosomal dominant manner, a common abnormality of which causes the development of peripheral nerve tumors consisting 60–85% Schwann cells and 10–20% fibroblasts with small amounts of pericytes, perineurial cells, mast cells, endothelial and smooth muscle cells.Citation12-Citation15 Some of NF1 patients develop to malignant peripheral nerve sheath tumor (MPNST) or low grade gliomas that are clinically resistant to conventional therapies. In addition, pheochromocytoma and myeloid leukemia are commonly seen in NF1 patients. In some cases, children with NF1 develop congenital skeletal dysplasias and learning disabilities.
PKC consists of more than 10 isoforms which are serine/threonine protein kinases.Citation16-Citation18 These isoforms differ in their structures, cellular functions and tissue distributions. The major isoforms, such as α and β, can be activated by both calcium and diacylglycerol (DAG), while other PKC subgroup (for example, δ or θ) is independent of calcium for their functions. The atypical PKC isozymes (ζ and λ/ν) require neither DAG nor calcium for their activation. Due to such differences, PKC isozymes are able to differentially regulate different cellular signaling pathways and dictate different biological outcomes, including apoptosis. Using small hairpin RNA (shRNA), we demonstrated that PKC α, β, or δ could be either pro- or anti-apoptotic, depending upon cell types or stimuli.Citation19-Citation21 It has also been reported that the stability of Nf1 protein was regulated by PKC and Nf1 deficient cells were highly sensitive to PKC inhibitors.Citation22 Recently, using genome-wide siRNA high-throughput screens, it revealed a diverse set of proteins whose depletion selectively impaired the viability of cells expressing aberrant or mutated ras.Citation23 Importantly, most of these proteins are kinases involving in the regulation of cell cycle, particularly mitosis.Citation23
Upon cellular insults, such as DNA damage, cell cycle checkpoints are activated to eliminate damaged DNA for being propagated, which prevents injured cells to enter next generation.Citation24-Citation26 Such elaborate signaling network during DNA damage is mainly regulated by 2 partially overlapped pathways: Chk1 and Chk2.Citation24-Citation26 The activation of these checkpoint regulators triggers cell cycle responses, damage repair and apoptosis. It is well known that mitotic catastrophe is often elicited when cells fail to finish or exit from the mitotic phase.Citation27,Citation28 In budding yeast, mitotic catastrophe was triggered after chromosomes were mis-segregated.Citation29-Citation31 In order to properly exiting from the mitosis and entering cytokinesis, cyclin B1 has to be degraded in time.Citation32-Citation34 A prolonged elevation of cyclin B1 expression was observed to be accompanied with a persistent mitotic arrest, resulting in mitotic catastrophe.Citation32-Citation34 It was also suggested that mitotic catastrophe is an important mechanism for eliminating cancerous or diseased cells, during which cell cycle checkpoint regulators play important roles.Citation35,Citation36 In the G2 and mitotic phases, the DNA damage checkpoint was realized by the phosphorylation and inactivation of Cdk1 or Cdc25C in a Chk1-dependent matter, whose function is essential for cell progression in the mitosis.Citation37-Citation39 However, the molecular mechanisms that link persistent mitotic arrest to the cell death machinery were not fully understood.
In this study, we identified that Chk1 was phosphorylated and activated in Nf1 deficient cells in the absence of PKC, accompanied by a persistent expression of cyclin B1, prolonged mitotic arrest and subsequent induction of apoptosis via mitotic catastrophe. We further demonstrated that these events occurred in HMG-treated Nf1 deficient cells were dependent upon Chk1. Overall, the study suggested that PKC is critical for maintaining homeostasis in the cellular environment controlled by aberrant Nf1 signaling.
Results
PKC activity was increased in Nf1 deficient cells
Cancer cells harboring an oncogenic or mutated ras appeared highly sensitive to chemical or genetic PKC inhibitors.Citation19-Citation22 However, it remained unclear whether Nf1 deficient cells would be susceptible to apoptosis in the absence of PKC. Therefore, human Nf1 deficient ST8814 cells were used in this study. The Nf1 effective domain gene was generated by PCR, and then inserted into the pBMN plasmid expression vector. The construct containing the Nf1 effective domain gene was stably transfected into ST8814 cells and designated as ST/NF1 cells. Subsequently, the activity of Ras in ST8814 or ST/Nf1 cells was measured, using the Active Ras Pull-Down and Detection kit. A high amount of the GTP bound Ras was detected in ST8814 cells (). In comparison, the active Ras was almost undetectable after ST8814 cells were transfected with Nf1 effective domain gene. The amount of the active Ras in ST8814 or ST/Nf1 cells did not change after the treatment of HMG (1-O-methyl-rac-glycerol, a PKC inhibitor) (data not shown). Akt and MAPK function downstream of Ras and have been implicated in the growth promotion under Nf1 deficient conditions.Citation40 Therefore, the phosphorylation status of these Ras effectors was analyzed by immunoblotting. A high level of the phosphorylation form of Akt or ERK1/2 was present in ST8814 cells, but absent in ST/Nf1 cells (). Again, the levels of the phosphorylation of these Ras effectors were not altered by the addition of HMG (data not shown). The activation of JNK or p38 in the cells was also tested. Neither JNK nor p38 was active in ST8814 or ST/Nf1 cells (data not shown).
Figure 1. Ras and PKC signaling in ST cells. (A) Cell lysates were extracted from ST8814 and ST/Nf1 cells and subjected to Ras Pull-Down assay. The even loadings of total proteins were normalized by Ras expression. (B) Cell lysates were prepared and then immunoblotted with the anti-phosphorylated Akt or ERK1/2 antibody. The even loadings of total proteins were normalized by Akt or ERK1/2 expression. (C) After the treatments of PMA, HMG, or both, cell lysates were subjected to immunoprecipitation with an anti-PKC antibody. PKC activity in the immunoprecipitates was then analyzed using a PKC enzymatic kit. The error bars represent SD from 5 independent experiments (n = 5, P values < 0.05).
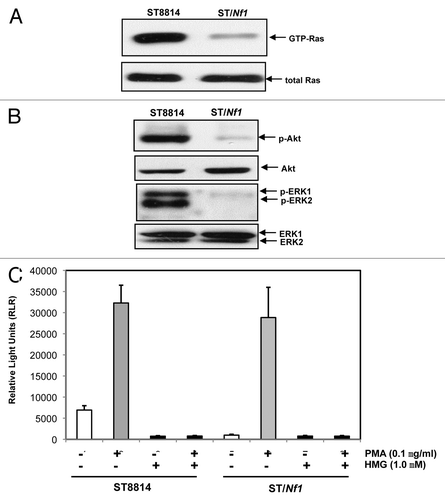
It was reported that Nf1 deficient cells were sensitive to PKC inhibitionCitation22 and cancer cells expressing mutated ras depended upon PKC for survival.Citation19-Citation21 These led us first to test PKC expression. A comparable amount of PKC was expressed in ST8814 or ST/Nf1 cells (data not shown). Subsequently, the cells were treated with PMA (phorbol myristate acetate, a PKC activator) or co-treated with PMA plus HMG, and the activity of this kinase was measured using a PKC kinase activity kit (). Interestingly, the activity of PKC was moderately high in untreated ST8814 cells, but was at the baseline level in untreated ST/Nf1 cells. As expected, PMA treatment dramatically upregulated PKC activity in both cells, and the addition of HMG alone had no impact on the kinase activity. HMG treatment completely suppressed PKC activity in untreated ST8814 cells as well as that induced by PMA in both ST8814 and ST/Nf1 cells. The data suggested that PKC activity was moderately, but persistently upregulated in Nf1 deficient cells.
HMG treatment exerted pro-apoptotic effect on Nf1 deficient cells in response to HMG treatment in vitro and in vivo
Next, we tested whether PKC inhibition conferred the susceptibility of Nf1 deficient cells to apoptosis. DNA fragmentation assay was conducted (). After being treated with HMG for 48 h, more than 40% of ST8814 cells underwent apoptosis. In comparison, very low numbers of ST/Nf1 cells, following HMG treatment, were apoptotic. Annexin V-FITC apoptotic detection assay was also conducted. A similar result was obtained (data not shown). It seemed that the inhibition of PKC sensitized the cells with Nf1 deficiency to apoptosis.
Figure 2. Suppression of PKC perturbed the growth of ST8814 cells in vitro and in vivo. (A) After the treatment with HMG (1.0 μM) for 48 h, DNA fragmentation assay was performed. The error bars are SD over 5 independent experiments (n = 5, * P < 0.05). (B) ST8814 cells were inoculated subcutaneously into the nude mice (4 mice per group). HMG was injected peritoneally right after the inoculation and subsequently administrated every 4 d. Thirty-two days later, the isolated tumors were weighted and data were plotted. The error bars represent SD (n = 4, *P values < 0.05). (C) The photo of the tumors isolated from untreated or HMG treated mice.
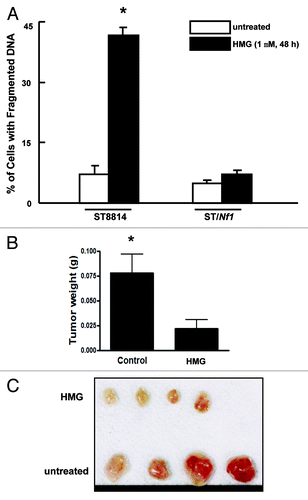
To further confirm the inhibitory effect of HMG on Nf1 deficient tumor growth in vivo, ST8814 cells were subcutaneously inoculated into nude mice. At the same time, the mice were injected with HMG peritoneally. Afterwards, the same concentration of HMG was administrated into the mice every 4 d. The tumor formation signs were noticed in untreated mice 10 d after the inoculation. However, the growth of the xenografted tumors was significantly delayed in HMG-treated mice. Thirty-two days after the inoculation, the mice were sacrificed and, tumors were isolated and weighted (). The untreated tumor messes were 3.5-folds bigger than those treated with HMG. The isolated tumors were also documented by the photograph (). The in vivo results further suggested that Nf1 deficiency was incompatible with loss of PKC for survival.
HMG-mediated cell death in ST8814 cells was via mitotic catastrophe
During conducting DNA fragmentation assay to detect apoptosis, we noticed that a large percentage of HMG-treated ST8814 cells seemingly accumulated in the mitotic phase of the cell cycle before undergoing apoptosis. This led us to analyze the cell cycle profiles of HMG-treated cells at the time point before undergoing apoptosis (). Thirty-two hours after HMG treatment, majority of ST8814 cells (more than 90%) accumulated in the G2/M phases. In comparison, much less percentage (<30%) of HMG-treated or untreated ST/Nf1 cells was presented in the mitotic phase after the treatment, suggesting that the majority of the treated ST/Nf1 cells, like the untreated cells, entered next cytokinesis. To further corroborate the link between the persistent mitotic arrest and mitotic catastrophe, ST8814 and ST/Nf1 cells, after being treated with HMG for 32 h, was stained with Giemsa dye (). Dramatically enlarged nuclei were observed in HMG-treated ST8814 cells, but not in the treated ST/Nf1 cells. Taken together with the data presented in , it seemed that apoptosis that occurred in HMG-treated Nf1 deficient cells was through a mitotic catastrophe. Since the introduction of the functional Nf1 effective domain gene into ST8814 cells prevented the occurrence of mitotic arrest and catastrophe, such reversion is probably dependent upon and specific for Nf1 function.
Figure 3. Persistent mitotic arrest and mitotic catastrophe occurred in HMG-treated ST8814 cells. (A) ST8814 or SNF02.2 cells were treated with HMG for 32 h, cell cycle analysis was performed using a flow cytometer. The percentages of the cells accumulated in the G2 and M phases were plotted (left panel). The error bars represent SD from 5 independent experiments (n = 5, * P values < 0.05). The DNA profiles of the untreated or HMG–treated ST8814 and ST/Nf1 cells were presented in the right panels. (B) Thirty-two hours after HMG treatment, the cells were stained with Giemsa dye and photos of the stained nuclei were taken. (C) With or without HMG treatment, cell lysates were prepared and subjected to immunoblotting with an anti-cyclin B1 antibody. The even loadings of total proteins were normalized by actin expression.
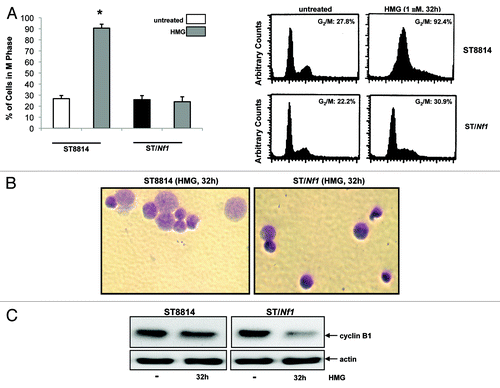
Cyclin B1 is regulated spatially during cell division. In particular, this cell cycle regulator is rapidly and temporally downregulated before cells exit from the mitotic phase and entering next cytokinesis.Citation32-Citation34 Because HMG treatment caused a persistent mitotic arrest of Nf1 deficient cells, it led us to examine the effect of this PKC inhibitor on cyclin B1 expression by immunoblot analysis (). The expression of cyclin B1 maintained at the similar level after ST8814 cells were treated with HMG for 32 h in comparison with that in untreated cells. The amount of cyclin B1 in ST/Nf1 cells was significantly decreased upon the treatment. Overall, the high expression of cyclin B1 in HMG-treated ST8814 cells was in a good agreement with that the same cells were unable to exit from the mitotic phase.
Chk1 was activated in HMG-induced mitotic arrest in Nf1 deficient cells
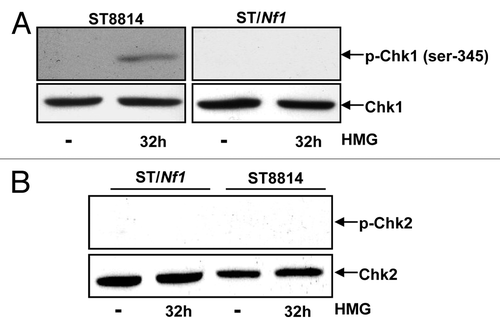
Chk1 and 2 are the key regulators in the regulation of cell cycle checkpoints.Citation24-Citation26 Since the Nf1 deficient cells delayed to exit from the mitotic phase of the cell cycle and cyclin B1 was persistently expressed in the cells following HMG treatment, the phosphorylation status of Chk1 () and 2 () kinases was examined in ST8814 or ST/Nf1 cells with or without HMG treatment, using the anti-phosphorylated S345-Chk1 or anti-phosphorylated-Chk2 antibody, respectively. Chk1 was phosphorylated at the serine-345 residue in HMG-treated ST8814 cells only. Chk2 phosphorylation was detected in neither ST8814 nor ST/Nf1 cells with or without HMG treatment.
Figure 4. Phosphorylation of Chk1 and 2 following HMG treatment. (A) After HMG treatment, The expression of the phosphorylated Chk1 in ST8814 and ST/Nf1 cells was analyzed by immunoblotting using the anti-phosphor-Chk1 (ser-345) antibody. The even loadings of total proteins were normalized by Chk1 expression. (B) After the treatment as described above, the expression of the phosphorylated Chk2 was analyzed by immunoblotting using the anti-phosphor-Chk 2 antibody. The even loadings of total proteins were normalized by Chk2 expression.
Subsequently, the knockdown effect of Chk1 by siRNA-Chk1on HMG-mediated mitotic exit delay was examined (). The knockdown of Chk1 by the siRNA was tested first. scRNA- or siRNA-Chk1 was transiently transfected into ST8814 cells. The cell lysates were prepared and subjected to immunoblotting with an anti-Chk1 antibody (). A similar amount of Chk1 was recognized by the antibody in control ST8814 cells or the cells transfected with scRNA-Chk1. Chk1 expression was almost undetected in the cells after the introduction of the siRNA. Subsequently, the DNA profiles were analyzed in HMG-treated cells transfected with scRNA- or siRNA-Chk1 (). Consistently, more than 90% of ST8814 cells still accumulated in the G2 and mitotic phases after the transfection of the scRNA. In contrast, the knockdown of Chk1 by siRNA-Chk1 relieved the major of HMG-treated ST cells (>70%) from arresting in the G2 and mitotic phases and, allowed these cells to enter next cytokinesis.
Figure 5. Effect of the knockdown of Chk1 on persistent mitotic arrest in HMG-treated ST8814 cells. (A) Forty-eight hours after transfected with scRNA- or siRNA-Chk1, Chk1 expression in ST8814 cells was analyzed by immunoblotting. The even loadings of total proteins were normalized by actin expression. (B) After the transfection of scRNA- or siRNA-Chk1 into ST6614 cells, ST8814 cells were treated with HMG for 32 h, cell cycle analysis was performed using a flow cytometer. The percentages of the cells accumulated in the G2 and M phases were plotted (left panel). The error bars represent SD from 5 independent experiments (n = 5, * P values < 0.05). The DNA profiles of the untreated or HMG–treated ST8814 cells transfected with either scRNA- or siRNA-Chk1 were presented in the right panels. (C) After knockdown of Chk1, cyclin B1 expression was examined by immunoblotting. The even loadings of total proteins were normalized by actin expression.
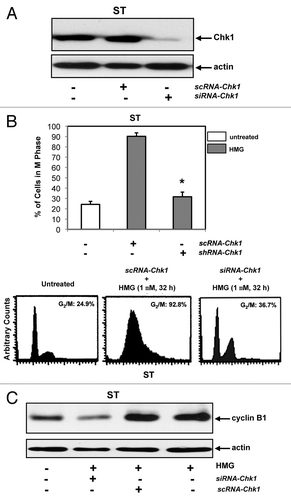
Since the levels of cyclin B1 in untreated and HMG-treated ST8814 cells were unchanged, the expression of cyclin B1 was tested after the knockdown of Chk1 (). The cells transfected with scRNA- or siRNA-Chk1 were treated with HMG for 32 h and then subjected to immunoblot analysis. A similar amounts of cyclin B1 were present in ST8814 cells treated with HMG and transfected with scRNA-Chk1 plus the treatment. However, the knockdown of Chk1 reversed the HMG-induced, persistent increases of cyclin B1 expression in ST8814 cells and much less amount (>2.5-folds) of this cell cycle regulator was detected.
HMG-induced mitotic catastrophe in Nf1 deficient cells is dependent upon Chk1 and Akt
We then tested the induction of apoptosis in Nf1 deficient cells in the absence of Chk1 or after the inhibition of Akt or MAPK (). ST8814 cells were transiently transfected with scRNA- or siRNA-Chk1, prior to the treatment of HMG or other inhibitors. Forty-eight hours later following PKC inhibition, Annexin V apoptotic assay was conducted. The magnitude of the induction of apoptosis in ST8814 cells transfected with siRNA-Chk1 was partially reduced in comparison with that occurred in the cells received HMG treatment only. The transfection of scRNA-Chk1 did not affect the magnitude of apoptosis induced by HMG. The effect of the inhibition of Akt or MAPK on the induction of apoptosis was also examined. The addition of KP372–1 (an Akt inhibitor), but not PD168393 (a MAPK inhibitor), partially blocked this HMG-mediated apoptotic process in ST8814 cells. The incomplete suppression of the apoptotic process indicated the involvement of other signaling pathways, besides Akt signaling, in this action.
Figure 6. Effect of suppression of Akt on the induction of apoptosis and Chk1 phosphorylation. (A) After the transfection of scRNA-, siRNA-Chk1, or treatment of Akt, ERK1/2 inhibitor, respectively, prior to the addition of HMG, annexin V analysis was performed. The error bars represent SD over 5 independent experiments (n = 5, * P values < 0.05). (B) The phosphorylation status of Chk1 in HMG-treated ST8814 cells, after the treatment of Akt or ERK1/2 inhibitor, was analyzed by immunoblotting. The even loadings were normalized by Chk1 expression.
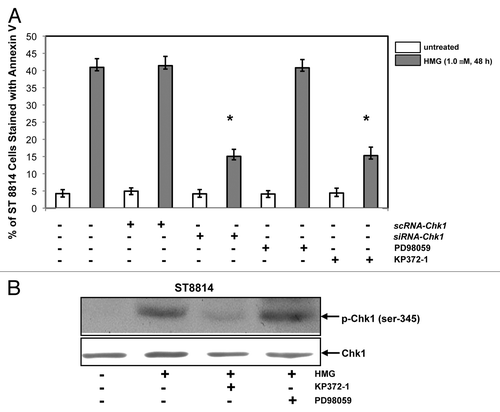
To further determine whether Akt played a role in Chk1 activation, the phosphorylation of Chk1 was tested in the presence of KP372–1 or PD98059 by immunoblot analysis (). The phosphorylated form of Chk1 was absent in HMG-treated ST8814 cells after the inhibition of Akt. However, the addition of PD98059 did not affect HMG-induced Chk1 phosphorylation. Overall, the data suggested that the suppression of endogenous PKC probably cooperated with Akt pathway to activate the Chk1-mediated, mitotic exit checkpoint in Nf1 deficient ST8814 cells, leading to apoptosis, possibly through mitotic catastrophe.
Discussion
Neurofibromatosis type 1 (NF1) is caused by mutations or deletions in Nf1, which render the high susceptibility to the development of benign or malignant tumors, such as neurofibrotoma, leukemia, or malignant peripheral nerve sheath tumors (MPNST) that are originated from neural crest-derived tissues.Citation41-Citation43 Because the underlying mechanisms of the high susceptibility of tumor formations in NF1 patients have not been fully understood, there is an urgent need for identifying molecular targets and further developing effective therapies to treat this disease, especially MPNST. Our current study demonstrated a potential, apoptotic signaling model, in which PKC activity appeared being upregulated in Nf1 deficient ST8814 cells and however, the inhibition of PKC sensitized the cells to apoptosis, possibly via mitotic catastrophe. Prior to the occurrence of apoptosis, the majority of the Nf1 deficient cells, after PKC inhibition, accumulated in the mitotic phase, which accompanied with the persistent expression of cyclin B1. Furthermore, the cell cycle checkpoint regulator Chk1 was activated. The inhibition of Chk1 or Akt blocked this apoptotic process. Collectively, our study suggested that the increase of PKC activity functions parallel with hyperactive Ras pathway to maintain the homeostasis in Nf1 deficient cells for survival. After the suppression of PKC signaling, Nf1 deficient cells can no longer keep high metabolic rates driven by aberrant Ras and an apoptotic crisis is initiated, which may be through activating a Chk1-mediated mitotic exit checkpoint. Our study also indicated that Akt may be one of downstream effectors of aberrant Ras and cooperates with loss of PKC for the activation of the mitotic checkpoint and further occurrence of mitotic catastrophe.
Under the condition of Nf1 deficiency, Ras activity is hyper-activated. In order to maintain the deregulated Ras signals or to keep high metabolic rates driven by aberrant Ras, a coordination of the Ras signaling with other parallel pathways may be essential for the survival of Nf1 deficient cells. Disruption of one or more of these signaling pathways would perturb the homeostasis under the Nf1 deficient conditions and further trigger an apoptotic crisis in the cells. Recently, targeting cooperating factors of aberrant Ras signaling has been viewed as an important strategy for developing anti-cancer therapies. It was reported that in the absence of PKC, aberrant Ras became unstable and subsequently triggered cell death program.Citation19-Citation21 However, little is known about whether PKC signaling is essential for Nf1 deficient cells to survive or if the blockade of PKC could sensitize the defect cells to apoptosis. In this study, we demonstrated the occurrence of a synthetic lethal interaction between mutated Nf1 and loss of PKC. In this lethal process, Chk1 was phosphorylated in Nf1 deficient cells after the treatment of HMG, and responsible for triggering a persistent mitotic arrest and subsequent cell death. The suppression of Akt not only abrogated Chk1 activation, but also dramatically decreased the magnitude of apoptosis in our experimental setting. However, it remains puzzling about the suggested linear relationship between Akt and Chk1 observed here, because Chk1 was not phosphorylated in untreated ST8814 cells and however, Akt, as the downstream of aberrant Ras, was being constitutively activated in the same cells. Previously, we showed that PKC δ functioned as a tumor suppression to promote apoptosis in cancer cells harboring oncogenic ras, when the conventional PKC isoforms were suppressed.Citation19 Therefore, it is possible that PKC δ is upregulated or activated and further acts in synergy with Akt to mediate Chk1 phosphorylation under Nf1 deficient condition. The investigation of Chk1 activation in our experimental setting is under way.
Genetic changes (such as the loss of the CDKN2A/B, encoded proteins of which are negative cell cycle regulators) could be detected at early stages of NF1.Citation44 In MPNSTs, the genes of p53 and other tumor suppressors are often mutated. In particular, under Nf1 deficient conditions, the expression and function of aurora kinase (a regulator of the mitosis) were upregulated to promote cell proliferation or migration, in a Ras-dependent fashion.Citation45 The inhibition of aurora was shown to induce mitotic catastrophe. It was also reported that Ras family protein Rac 1 was functioning during the G2 and mitotic phases to promote cell division.Citation46 These data all suggested the involvement of Ras or its family members in the regulation of the G2 and mitotic phases. Thus, the focus on the mitotic phase and its regulators may shed a light for developing new strategies for NF1 therapeutics, especially for MPNST.
Mitotic catastrophe is a well-known phenomenon and often occurs in cancer cells treated with genotoxic or radiation reagents.Citation32-Citation34 Many studies demonstrated that upon genotoxic insults, cells (including mammalian cells) were accumulated in the mitosis by the delay of mitotic exit.Citation39 The connection between mitotic DNA damage checkpoint and mitotic catastrophe is well established in irradiated cancer cells.Citation36 It was reported that the mitotic exit checkpoint was activated by Chk1 in the cancer cells, followed by the occurrence of mitotic catastrophe.Citation39 Our current study indicated that the inhibition of PKC appeared targeting a mitotic checkpoint, which further caused Nf1 deficient cells to collapse at the mitosis. Since PKC activity was upregulated in Nf1 deficient ST8814 cells, it is likely that PKC cooperates with aberrant Ras to maintain the homeostasis in the cells. Once PKC activity was inhibited, hyperactive Ras functions through Akt and other signaling pathway to activate the Chk1-dependent mitotic exit checkpoint, leading to mitotic catastrophe in Nf1 deficient cells. The underlying mechanisms of how this Chk1-dependent, mitotic exit checkpoint is activated in Nf1 deficient cells remain to be further investigated. In budding yeast, the mitotic exit checkpoint is operated by Pds1 through metaphase arrest.Citation47 This checkpoint could also be activated by Bub2, a negative regulator of the mitotic exit.Citation48 It is conceivable that an equivalent factor, like Bub2, is controlled by Akt and Chk1 for the activation of the mitotic exit checkpoint in our experimental setting. Furthermore, the knockdown of Chk1 or inhibition of Akt only partially blocked the magnitude of apoptosis, which indicates the involvement of multiple apoptotic pathways in our experimental setting.
Studies showed that the treatment with PKC inhibitors could induce apoptosis in various types of cells overexpressing oncogenic v-ras.Citation19-Citation21 Here, we identified that the suppression of PKC was able to induce apoptosis in human Nf1 deficient cells, via eliciting persistent mitotic arrest and further mitotic catastrophe, but not in the same cells expressing a normal Nf1 functional domain gene. The involvement of PKC in the regulation cell cycle progression was well documented.Citation49,Citation50 The treatment of PKC inhibitors appeared promoting re-replication without the completion of cell cycle, leading to the deregulation of the mitotic phase and the generation of cells with polyploidies.Citation51,Citation52 In this study, we showed that after HMG treatment, the size of some of ST8814 cells were significantly enlarged (), before the onset of mitotic catastrophe. Indeed, the percentage of HMG-treated ST8814 cells with polyploidies was dramatically increased in comparison with that of HMG-treated ST/Nf1 cells (data not shown). All the information indicated that targeting PKC may be an ideal strategy for developing new therapeutics to treat MPNST patients. Importantly, the inhibition of PKC is not harmful for surrounding normal cells or tissues.Citation19-Citation21
In summary, our study shows that the suppression of PKC specifically induces the cells with Nf1 deficiency to undergo apoptosis, suggesting that hyperactive Ras is the possible player in the regulation of this apoptotic process. PKC and Nf1 are important intracellular signal transducers for cell differentiation and proliferation. Nf1 defects alone are compatible with cell viability. However, the suppression of PKC severely perturbs crucial survival signaling pathways in Nf1 deficient cells, which elicited an apoptotic crisis through the Chk1-dependent, mitotic exit checkpoint. With increasing attention in targeting parallel or downstream signaling of aberrant Ras pathway for cancer treatment, our study provides the new information for developing therapeutic strategies that are able to preferentially kill Nf1 deficient tumor cells at clinically achievable doses.
Materials and methods
Cells and reagents
Human NF1 deficient ST8814 cells were obtained from ATCC. The cells were cultured in Dulbecco Modified Eagles medium supplemented with 10% heat-inactivated Fetal Bovine Serum (Atlanta Biologicals), 100 units/ml penicillin, 100 µg/ml Streptomycin. ST6614 cells were stably transfected with a plasmid pBMN vector (Invitrogen) carrying the Nf1 effective domain and maintained in the growth medium containing 400 μg/ml of G418 (neomycin) (Life Technlogies). HMG and PD98059 were purchased from EMD Millipore and KP372–1 was from Echelon. Antibodies were purchased from BD Biosciences. The siRNA-Chk1 was purchased from Origene.
Ras activation assay
Active Ras Pull-Down and Detection kit (Thermo. Scientific) was used. Positive control was generated by treating lysate extracted from the cells with GTPγS to activate Ras.
PKC activity assay
The activity of PKC was analyzed using thPKC light HTS protein kinase assay kit (Lonza Rocklnad). Cell lysates were immunoprecipitated with an anti-PKC antibody. Subsequently, the immunoprecipitates were incubated with PKC substrate peptide and adenosine triphosphate (ATP) in the kinase buffer. The intensity of the luminescence in the samples was determined by a microplate luminometer.
Immunoprecipitation and immunoblot analyses
After treatments, cells were lysed and immunoprecipited with antibodies. Subsequently, the precipitates pulled down by the antibodies were separated by SDS-PAGE gel and transferred to nitrocellulose. After blocking with 5% non-fat milk for 1 h at room temperature, the nitrocellulose was probed with antibodies and then visualized by chemilluminescence (Perkin-Elmer).
Flow cytometry analysis
Flow cytometry analyses for cell cycle profile and fragmented DNA were performed using a FACScan machine (BD Biosciences). The data analysis was performed using the Cell-Fit software program (BD Biosciences). Cell-Fit receives data from the flow cytometer and provides real-time statistical analysis, computed at one second intervals, and also discriminates doublets or adjacent particles. Following treatments, cells were harvested and then fixed in 70% cold ethanol. Afterwards, cells were stained with 0.1 mg/ml propidium iodide containing 1.5 ug/ml of RNase. DNA profiles of cells or sub-G0-G1 DNA contents of cells were then analyzed by the FACScan machine.
Annexin V-FITC apoptosis detection assay
After treatments, cells were prepared and stained with Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences) according to manufacturer’s instructions. Subsequently, the samples were analyzed by a flow cytometer.
Xenograft assay
Nude mice at ages of 4–6 wk were used for the xenograft assay. ST8814 cells (5 × 106) in 100 μl of 1 × PBS were inoculated into the right flank area of each mouse. Four mice were served as the untreated control. For the inhibition of tumor formation, 4 mice were injected peritoneally with HMG (30 mg/kg in 100 μl of 1 × PBS) right after the inoculation and subsequently administrated the same amount of HMG every 3 d. The sizes of the tumors were measured routinely. Thirty-two days later, the mice were sacrificed and the tumors were isolated and weighted.
Giemsa staining assay
After HMG treatment for 32 h, the cells were fixed on slides in methanol for 30 min and stained with a freshly-made 10% Giemsa staining solution for 30 min at room temperature. The slides were rinsed with distilled water and then thoroughly dried.
Statistical analysis
Averages and standard deviations of the results of the experiments were computed. Standard deviations are displayed as error bars in the figures. A Student t test was used and a P value of < 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Luo (Boston University School of Medicien, MA) for providing reagents and help suggestions with the project. This study is supported by NIH R01CA153354 grant (to C.C.) and the South Korean Government MEST 2012-00057555 (to S.-H.K.). Dr X. Zhou is supported by the research grant from the Chinese Science Foundation. Dr S.-H. Kim is supported by the grant from the South Korean Government MEST (2012-0005755).
References
- Weiss B, Bollag G, Shannon K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet 1999; 89:14 - 22; http://dx.doi.org/10.1002/(SICI)1096-8628(19990326)89:1<14::AID-AJMG5>3.0.CO;2-4; PMID: 10469432
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990; 63:851 - 9; http://dx.doi.org/10.1016/0092-8674(90)90151-4; PMID: 2121371
- Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P, Cawthon RM, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990; 63:843 - 9; http://dx.doi.org/10.1016/0092-8674(90)90150-D; PMID: 2121370
- Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990; 62:599 - 608; http://dx.doi.org/10.1016/0092-8674(90)90024-9; PMID: 2116237
- Thomas L, Richards M, Mort M, Dunlop E, Cooper DN, Upadhyaya M. Assessment of the potential pathogenicity of missense mutations identified in the GTPase-activating protein (GAP)-related domain of the neurofibromatosis type-1 (NF1) gene. Hum Mutat 2012; 33:1687 - 96; http://dx.doi.org/10.1002/humu.22162; PMID: 22807134
- Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992; 356:713 - 5; http://dx.doi.org/10.1038/356713a0; PMID: 1570015
- DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 1992; 69:265 - 73; http://dx.doi.org/10.1016/0092-8674(92)90407-4; PMID: 1568246
- Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 1996; 12:144 - 8; http://dx.doi.org/10.1038/ng0296-144; PMID: 8563751
- Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell 2001; 104:593 - 604; http://dx.doi.org/10.1016/S0092-8674(01)00245-8; PMID: 11239415
- Arun D, Gutmann DH. Recent advances in neurofibromatosis type 1. Curr Opin Neurol 2004; 17:101 - 5; http://dx.doi.org/10.1097/00019052-200404000-00004; PMID: 15021234
- Gottfried ON, Viskochil DH, Fults DW, Couldwell WT. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery 2006; 58:1 - 16, discussion 1-16; http://dx.doi.org/10.1227/01.NEU.0000190651.45384.8B; PMID: 16385324
- Gutmann DH, Parada LF, Silva AJ, Ratner N. Neurofibromatosis type 1: modeling CNS dysfunction. J Neurosci 2012; 32:14087 - 93; http://dx.doi.org/10.1523/JNEUROSCI.3242-12.2012; PMID: 23055477
- Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet 1999; 89:1 - 6; http://dx.doi.org/10.1002/(SICI)1096-8628(19990326)89:1<1::AID-AJMG3>3.0.CO;2-8; PMID: 10469430
- Hyman SL, Shores A, North KN. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 2005; 65:1037 - 44; http://dx.doi.org/10.1212/01.wnl.0000179303.72345.ce; PMID: 16217056
- Kalamarides M, Acosta MT, Babovic-Vuksanovic D, Carpen O, Cichowski K, Evans DG, et al. Neurofibromatosis 2011: a report of the children’s tumor foundation annual meeting. 2012; Acta Neuropathol: 123: 369-80.
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 1995; 9:484 - 96; PMID: 7737456
- Spitaler M, Cantrell DA. Protein kinase C and beyond. Nat Immunol 2004; 5:785 - 90; http://dx.doi.org/10.1038/ni1097; PMID: 15282562
- Reyland ME. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front Biosci (Landmark Ed) 2009; 14:2386 - 99; http://dx.doi.org/10.2741/3385; PMID: 19273207
- Zhu T, Tsuji T, Chen C. Roles of PKC isoforms in the induction of apoptosis elicited by aberrant Ras. Oncogene 2010; 29:1050 - 61; http://dx.doi.org/10.1038/onc.2009.344; PMID: 19838205
- Zhu T, Chen L, Du W, Tsuji T, Chen C. Synthetic lethality induced by loss of PKC delta and mutated Ras. Genes Cancer 2010; 1:142 - 51; http://dx.doi.org/10.1177/1947601909360989; PMID: 21031151
- Shen L, Kim SH, Chen CY. Sensitization of human pancreatic cancer cells harboring mutated K-ras to apoptosis. PLoS One 2012; 7:e40435; http://dx.doi.org/10.1371/journal.pone.0040435; PMID: 22848379
- McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SM, Nghiemphu P, Liau LM, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 2009; 16:44 - 54; http://dx.doi.org/10.1016/j.ccr.2009.05.009; PMID: 19573811
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009; 137:835 - 48; http://dx.doi.org/10.1016/j.cell.2009.05.006; PMID: 19490893
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433 - 9; http://dx.doi.org/10.1038/35044005; PMID: 11100718
- Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 2009; 21:245 - 55; http://dx.doi.org/10.1016/j.ceb.2009.01.018; PMID: 19230643
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179 - 204; http://dx.doi.org/10.1016/j.molcel.2010.09.019; PMID: 20965415
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12:385 - 92; http://dx.doi.org/10.1038/nrm3115; PMID: 21527953
- Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007; 6:70 - 4; http://dx.doi.org/10.4161/cc.6.1.3682; PMID: 17245109
- Booher R, Beach D. Involvement of a type 1 protein phosphatase encoded by bws1+ in fission yeast mitotic control. Cell 1989; 57:1009 - 16; http://dx.doi.org/10.1016/0092-8674(89)90339-5; PMID: 2544292
- Löffler H, Rebacz B, Ho AD, Lukas J, Bartek J, Krämer A. Chk1-dependent regulation of Cdc25B functions to coordinate mitotic events. Cell Cycle 2006; 5:2543 - 7; PMID: 17106257
- Jensen S, Johnston LH. Complexity of mitotic exit. Cell Cycle 2002; 1:300 - 3; http://dx.doi.org/10.4161/cc.1.5.142; PMID: 12461287
- Dumitrescu TP, Saunders WS. The FEAR Before MEN: networks of mitotic exit. Cell Cycle 2002; 1:304 - 7; http://dx.doi.org/10.4161/cc.1.5.147; PMID: 12461288
- Lee TH, Kirschner MW. An inhibitor of p34cdc2/cyclin B that regulates the G2/M transition in Xenopus extracts. Proc Natl Acad Sci U S A 1996; 93:352 - 6; http://dx.doi.org/10.1073/pnas.93.1.352; PMID: 8552637
- Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol 2007; 8:894 - 903; http://dx.doi.org/10.1038/nrm2276; PMID: 17912263
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23:2825 - 37; http://dx.doi.org/10.1038/sj.onc.1207528; PMID: 15077146
- Eriksson D, Löfroth PO, Johansson L, Riklund KA, Stigbrand T. Cell cycle disturbances and mitotic catastrophes in HeLa Hep2 cells following 2.5 to 10 Gy of ionizing radiation. Clin Cancer Res 2007; 13:5501s - 8s; http://dx.doi.org/10.1158/1078-0432.CCR-07-0980; PMID: 17875782
- Piwnica-Worms H. Reversible phosphorylation and mitotic control. J Lab Clin Med 1996; 128:350 - 4; http://dx.doi.org/10.1016/S0022-2143(96)80005-9; PMID: 8833882
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997; 277:1497 - 501; http://dx.doi.org/10.1126/science.277.5331.1497; PMID: 9278511
- Huang X, Tran T, Zhang L, Hatcher R, Zhang P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc Natl Acad Sci U S A 2005; 102:1065 - 70; http://dx.doi.org/10.1073/pnas.0409130102; PMID: 15650047
- Sandsmark DK, Zhang H, Hegedus B, Pelletier CL, Weber JD, Gutmann DH. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer Res 2007; 67:4790 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-4470; PMID: 17510408
- Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev 1994; 8:1019 - 29; http://dx.doi.org/10.1101/gad.8.9.1019; PMID: 7926784
- Parkin B, Ouillette P, Wang Y, Liu Y, Wright W, Roulston D, Purkayastha A, Dressel A, Karp J, Bockenstedt P, et al. NF1 inactivation in adult acute myelogenous leukemia. Clin Cancer Res 2010; 16:4135 - 47; http://dx.doi.org/10.1158/1078-0432.CCR-09-2639; PMID: 20505189
- Bottillo I, Ahlquist T, Brekke H, Danielsen SA, van den Berg E, Mertens F, Lothe RA, Dallapiccola B. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol 2009; 217:693 - 701; http://dx.doi.org/10.1002/path.2494; PMID: 19142971
- Beert E, Brems H, Daniëls B, De Wever I, Van Calenbergh F, Schoenaers J, Debiec-Rychter M, Gevaert O, De Raedt T, Van Den Bruel A, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer 2011; 50:1021 - 32; http://dx.doi.org/10.1002/gcc.20921; PMID: 21987445
- Patel AV, Eaves D, Jessen WJ, Rizvi TA, Ecsedy JA, Qian MG, Aronow BJ, Perentesis JP, Serra E, Cripe TP, et al. Ras-driven transcriptome analysis identifies aurora kinase A as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin Cancer Res 2012; 18:5020 - 30; http://dx.doi.org/10.1158/1078-0432.CCR-12-1072; PMID: 22811580
- Michaelson D, Abidi W, Guardavaccaro D, Zhou M, Ahearn I, Pagano M, Philips MR. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol 2008; 181:485 - 96; http://dx.doi.org/10.1083/jcb.200801047; PMID: 18443222
- Tinker-Kulberg RL, Morgan DO. Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev 1999; 13:1936 - 49; http://dx.doi.org/10.1101/gad.13.15.1936; PMID: 10444592
- Cooper JA, Nelson SA. Checkpoint control of mitotic exit--do budding yeast mind the GAP?. J Cell Biol 2006; 172:331 - 3; http://dx.doi.org/10.1083/jcb.200512153; PMID: 16431930
- Barboule N, Lafon C, Chadebech P, Vidal S, Valette A. Involvement of p21 in the PKC-induced regulation of the G2/M cell cycle transition. FEBS Lett 1999; 444:32 - 7; http://dx.doi.org/10.1016/S0014-5793(99)00022-8; PMID: 10037143
- Besson A, Yong VW. Involvement of p21(Waf1/Cip1) in protein kinase C alpha-induced cell cycle progression. Mol Cell Biol 2000; 20:4580 - 90; http://dx.doi.org/10.1128/MCB.20.13.4580-4590.2000; PMID: 10848585
- Baatout S, Chatelain B, Staquet P, Symann M, Chatelain C. Interaction between protein kinase C and actin in megakaryocyte polyploidization. Anticancer Res 1999; 19:5B 4193 - 8; PMID: 10628374
- Williams CM, Harper MT, Poole AW. PKCα negatively regulates in vitro proplatelet formation and in vivo platelet production in mice. Platelets 2014; 25:62 - 8; http://dx.doi.org/10.3109/09537104.2012.761686; PMID: 23402219