
Abstract
Sorting of membrane proteins within the secretory pathway of eukaryotic cells is a complex process involving discrete sorting signals as well as physico-chemical properties of the transmembrane domain (TMD). Previous work demonstrated that tail-anchored (TA) protein sorting at the interface between the Endoplasmic Reticulum (ER) and the Golgi complex is exquisitely dependent on the length and hydrophobicity of the transmembrane domain, and suggested that an imbalance between TMD length and bilayer thickness (hydrophobic mismatch) could drive long TMD-containing proteins into curved membrane domains, including ER exit sites, with consequent export of the mismatched protein out of the ER. Here, we tested a possible role of curvature in TMD-dependent sorting in a model system consisting of Giant Unilamellar Vesicles (GUVs) from which narrow membrane tubes were pulled by micromanipulation. Fluorescent TA proteins differing in TMD length were incorporated into GUVs of uniform lipid composition or made of total ER lipids, and TMD-dependent sorting and diffusion, as well as the bending rigidity of bilayers made of microsomal lipids, were investigated. Long and short TMD-containing constructs were inserted with similar orientation, diffused equally rapidly in GUVs and in tubes pulled from GUVs, and no difference in their final distribution between planar and curved regions was detected. These results indicate that curvature alone is not sufficient to drive TMD-dependent sorting at the ER-Golgi interface, and set the basis for the investigation of the additional factors that must be required.
Introduction
The ability of biological membranes to alternate between planar and highly curved geometries is of fundamental importance for essential biological processes, including vesicular traffic, cell division, and generation of the complex architecture of organelles. Most phospholipids of biological membranes are cylinder-shaped and therefore favor a planar geometry of the bilayer. Thus, membrane curvature in cells must be induced by active mechanisms, which often co-exist and cooperate. These include the action of membrane-bending proteins, traction by the cytoskeleton, integral membrane protein crowding, enzymatic modifications of phospholipids, asymmetric distribution of lipids between the two monolayers, and line tension between lipid domains of different thickness (reviewed in Refs Citation1–Citation3).
Inherent to membrane bending is the establishment of compositional differences between curved and planar regions. For instance, lipid microdomain formation favors bilayer bending, and wedge-shaped proteins or cone-shaped lipids are necessarily concentrated in the curved domains that they induce.Citation1,Citation2,Citation4 The cause-effect relationship between domain formation and curvature is reciprocal, i.e., domain formation can induce curvature, but the opposite is also true: pulling tubes out of planar bilayers composed of lipid mixtures can result in lipid sorting between the tubular and the flat zonesCitation5,Citation6 and the recruitment of peripheral curvature-sensing proteins may alter the composition of the curved domain.Citation7 Thus, in vesicular membrane traffic, membrane curvature on the one hand generates the carriers needed for cargo transport and on the other, it may play a role in concentrating membrane proteins and lipid cargoes into these carriers.
Recruitment of integral membrane proteins to curved, budding vesicles is an essential step in their transport along the secretory and endocytic pathways. This recruitment is generally thought to occur via direct binding of sorting signals on cargo membrane proteins to components of the cytosolic coat that drives the budding process.Citation8,Citation9 However, transmembrane protein sorting is dictated not only by discrete sorting signals, but also by the physico-chemical characteristics of the transmembrane domains (TMD).Citation10-Citation15 Studies in model systems and in cells as well as bioinformatic analyses indicate that the mismatch between TMD length and lipid bilayer thickness, known as the hydrophobic mismatch, plays a prominent role in TMD-dependent sorting.Citation16 In general, proteins with longer TMDs are exported from their initial site of insertion, the Endoplasmic Reticulum (ER), to downstream compartments of the secretory pathway, endowed with thicker bounding membranes. Although in some cases cargo receptors sensitive to TMD length may play a role in this TMD-dependent sorting process,Citation17,Citation18 the hydrophobic mismatch alone might drive recruitment of cargo into curved, budding vesicle domains. For instance, TMD-dependent sorting of C-tail-anchored proteins (TA proteins, reviewed in ref. Citation19) at the ER-Golgi interface does not depend on the recently characterized erv14 receptor that selects its cargo at ER exit sites (ERES).Citation18 Rather, the results of our imaging studies with model TA proteins differing in TMD length were compatible with the idea that TMD-dependent sorting at the ER-Golgi interface is driven by simple TMD partitioning within the bilayer and not by protein-receptor interactions.Citation12
Of the two fluorescently-tagged TA proteins (FP) that we investigated, the first (FP-22, containing a 22 residue-long TMD) is exported from the ER to reach the plasma membrane, while the second (FP-17, containing a 17 residue-long TMD) is a resident of the ER.Citation20 Immediately after synthesis, when both proteins are still in the ER, we found that FP-22 is restricted to ER tubules and excluded from sheets; in addition, it is recruited to ERES, from which it rapidly exchanges into the bulk ER. Instead, FP-17 is distributed throughout the entire ER, but impoverished in ERES.Citation12 The exclusion of the long TMD from flat cisternal domains of the ER and its segregation in curved domains are difficult to reconcile with a bona fide protein-receptor interaction and led us to hypothesize that FP-22’s long mismatched TMD might drive partitioning without the involvement of direct protein-protein interactions. This phenomenon could underlie the sorting of FP-22 into ERES and subsequent transport to the plasma membrane.
Here, we have investigated whether curvature alone can induce TMD-dependent sorting. To this aim, we have developed a model system consisting of Giant Unilamellar Vesicles (GUV) reconstituted with either FP-17 or -22, from which tubes are pulled out by molecular motorsCitation21,Citation22 or by optical tweezers.Citation23 Our approach allows us to investigate partitioning of transmembrane proteins between bilayer domains with different geometries and is applicable to different model proteins and lipid mixtures.
Results and Discussion
Generation and characterization of GUVs containing integrated TA proteins
The experimental plan of this work is illustrated in . Large Unilamellar Vesicles (LUVs), composed either of the abundant ER lipid palmitoyl-oleyl-phosphatidylcholine (POPC) or of total ER lipids, plus the integrated TA constructs, were produced by detergent dialysis. The short TMD of FP-17 is expected to match the bilayer thickness of the resulting vesicles, while the long TMD of FP-22 is predicted to have a positive mismatch (i.e., the TMD is longer than the thickness of the hydrophobic core of the lipid bilayer, see Discussion in ref. Citation12. The proteoliposomes were then subjected to electroformation to generate GUVs containing the inserted proteins. These GUVs can be micromanipulated to pull out nanotubes (reviewed in ref. Citation24), enabling the investigation of the effect of curvature on TMD-dependent sorting.
Figure 1. Experimental setup. (A) Schematic representation of TA constructs expressed in cells (FP-17 and FP-22) or used for production of recombinant proteins (recombinant FP-17 and FP-22). The Fluorescent Protein (FP) is either mEGFP or tdTomato. (B) Flowchart of the experiments (blue: lipids, red: detergent).
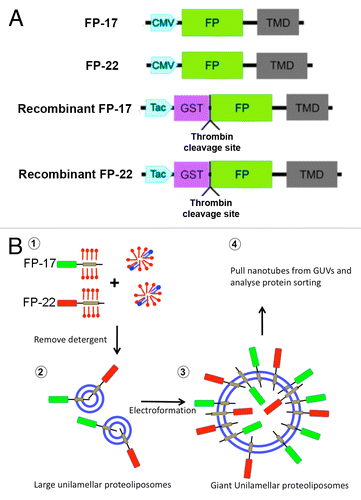
TA constructs consisting of a fluorescent cytosolically exposed domain (tdTomato -Citation25 - or mEGFP) followed by a TMD of either 17 or 22 residues (see ref. Citation20, for the sequence of the TMD and linkers) were cloned under the CMV promoter, or fused to GST under the Tac promoter to produce the recombinant proteins (). We named the encoded proteins mEGFP- or Tomato- 17 or 22, or simply FP- (Fluorescent Protein) 17 or 22 when referring to the short or long TMD constructs regardless of the fluorophore. We previously demonstrated that, when expressed in cells, the 22-long residue TMD allows escape of different GFP-derived variants from the ER to the plasma membrane.Citation12,Citation20 Since tdTomato is based on another fluorophore,Citation25 we first verified whether this construct behaves similarly to the EGFP-based constructs when expressed in cells. Like mEGFP-17 and 22 (, left panels), transfected Tomato-17 showed a typical ER staining pattern, whereas Tomato-22 was mainly detected on the cell surface (, right panels).
Figure 2. Characterization of the fluorescent proteins used in this study. (A) Distribution of the proteins after transfection into cultured cells. CV1 cells were fixed and imaged 24 h after transfection with FP-17 or FP-22. Scale bar, 10 μm. (B) SDS-PAGE analysis of the recombinant proteins. The same fluorescent variants as in (A) were purified from bacteria with the GST-fusion protein system. The asterisks and crosses indicate the bands corresponding to full-length FP-17 and FP-22, respectively (identified also by Western Blot in the case of mEGFP, see ). The lower bands are due to degradation occurring during the purification procedure. The arrowhead indicates the bacterial DnaK chaperon. Numbers on the left indicate the position and molecular weight (in kDa) of size markers.
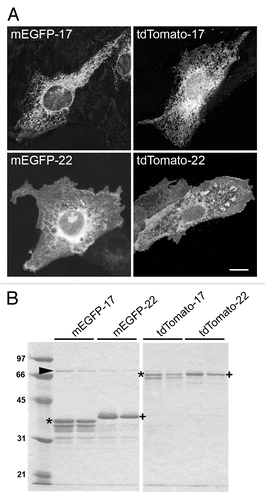
We then produced the four recombinant proteins in bacteria, and purified them by affinity chromatography followed by thrombin cleavage. SDS-PAGE analysis revealed a major band with the expected apparent molecular mass for each recombinant protein (). A number of minor bands of lower molecular weight, corresponding to degradation products, were also detected, especially in the case of the FP-17 samples.
The recombinant proteins were incorporated into LUVs composed either exclusively of POPC, an abundant ER lipid, or of total rat liver microsomal lipids. Analysis of negatively stained specimens by transmission EM revealed fairly homogenous preparations of spherical vesicles with an average diameter of 50 nm ().
Figure 3. Analysis of TA protein-containing proteoliposomes. (A) Transmission EM analysis of negatively stained proteoliposomes reconstituted with the indicated TA proteins. Scale bar, 500 nm. (B) Alkaline sucrose gradient analysis of LUVs reconstituted with FP-17 and 22. Reconstituted proteoliposomes (left) or the purified proteins without lipids (right) were treated with sodium carbonate. After flotation on a discontinuous sucrose gradient, fractions were collected and analyzed by Western Blot with an anti-GFP antibody. In proteoliposomes, the full-length form of both FP-17 and FP-22 float to the top of the gradient (top, light fractions), whereas the two proteins alone remain in the load zone (bottom, heavy fractions).
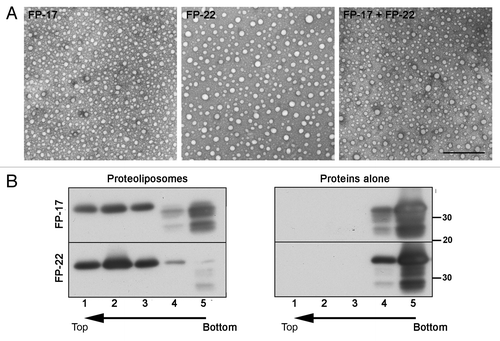
The bona fide integration of FP-17 and 22 into the lipid bilayer of the LUVs was analyzed by a stringent biochemical assay, consisting of carbonate extraction of the proteoliposomes followed by flotation on alkaline sucrose gradients. When the two proteins alone (not incorporated into liposomes) were analyzed, they were recovered exclusively in the load zone (bottom two fractions, , right panel) as expected. Instead, when reconstituted in proteoliposomes, the same two proteins were nearly quantitatively recovered in the top gradient fractions, in which the low density liposomes are found (, left panel), indicating their tight integration in the lipid bilayer. Notably, all the degradation products (see ) remained in the load zone, suggesting that even partial loss of the hydrophobic tail resulted in the incapacity of the constructs to insert into the bilayer.
The reconstituted LUVs were used to generate GUVs (Methods and Fig. S1). Confocal analysis of these GUVs revealed a homogeneous lateral distribution of both FP-17 and FP-22 ( and ). To determine the orientation of the FPs within the GUV membrane, we injected proteinase K (PK) into the GUV–containing chamber; as the bilayer is impermeable to macromolecules, PK is expected to digest only the portion of TA proteins exposed to the outside of the vesicles. As illustrated in and Videos S1 and S2, after PK injection, vesicles containing either tdTomato-17 or -22 first displayed constant fluorescence that then decreased over a period of ~1 min to about 50% of the initial value; fluorescence then remained constant during subsequent image acquisition. The initial (variable) constant phase can be attributed to the time it takes the PK to diffuse to the GUVs from the site of injection. Given the difficulty of capturing the initial stage of the digestion, we compared the fluorescence intensity of a large number of GUVs without addition of PK and after PK digestion at steady-state. As shown in , FP-17 and -22 showed similar behavior, with a reduction of fluorescence of ~50% at 10 min after addition of PK. These results suggested that the FPs are oriented randomly in the two orientations. To confirm this, once the steady-state effect of PK was reached, we added Triton X100 at low concentration (1/10000) to permeabilize the GUVs ( and Video S3). Permeabilization first induced a decrease in membrane fluorescence concomitant with an increase in the fluorescence of the interior of the GUVs in parallel with large membrane shape fluctuations due to a drop in membrane tension, and then a complete loss of both membrane and internal fluorescence. This sequence of events indicates that PK crosses the bilayer and cuts the internally oriented FPs that can then diffuse inside the GUV before being fully digested by PK and/or diffusing outside of the GUV through larger pores in the membrane as the permeabilization by Triton X100 progresses.
Figure 4. Orientation of TA proteins within GUV membranes. (A) Sequences of confocal images taken in the equatorial plane of tdTomato-17 (top) and tdTomato -22 (bottom) GUVs at the indicated times after addition of Proteinase K (PK). The graphs on the right show the fluorescence intensity as a function of time. The fluorescence drop occurs at different times depending on the diffusion of PK within the observation chamber. Arrows point to the time points at which the images were taken. See supplementary materials for the full time-lapse movies (Videos S1 and S2). (B) Quantification of the fluorescence drop due to digestion by PK of the FPs oriented toward the outside of the GUVs reconstituted with the tdTomato-17, tdTomato-22 or tdTomato-17/mGFP-22 TA proteins. Data were normalized by the value before PK addition. n = 62, 51 and 26 for tdTomato-17, tdTomato-22 and tdTomato-17/mGFP-22 respectively. ***: P < 10−6 in Student’s t test for the difference between PK-exposed and not exposed GUVs. (C) Sequences of confocal images taken in the equatorial plane of a tdTomato-17/mGFP-22 GUVs at the indicated times after addition of Triton-X100. The detergent was added 12 min after exposure of the GUVs to PK. The graph on the right shows the fluorescence intensity inside the GUV as a function of time. Arrows point to the time points at which the images were taken. See supplementary materials for the full time-lapse movie (Video S3). Scale bars in (A) and (C), 10 μm.
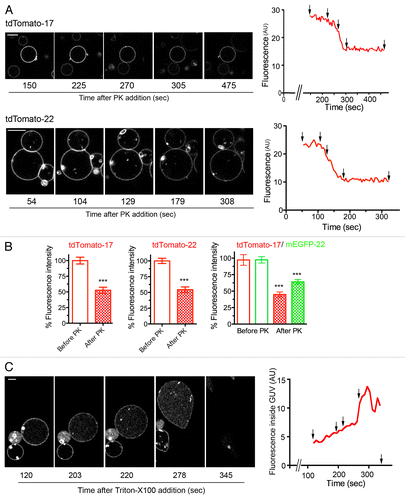
Figure 5. Distribution of TA proteins in GUVs and in tubular networks pulled from GUVs by kinesin motors. (A) Confocal images taken in the equatorial plane (top) and at the coverslip surface (bottom) where the tubes are generated. Images in the two planes were acquired with the same illumination and acquisition settings. (B) Quantification method used to analyze the distribution of FP-17 and FP-22 in GUVs and in nanotubes (see Methods). Scale bars in (A) and (B), 10 μm.
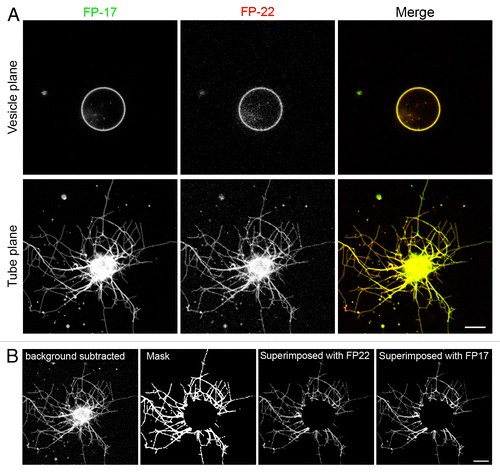
Analysis of curvature-driven sorting in tube networks pulled by kinesin motors
To pull out nanotubes from the GUVs, we applied a technique developed by Roux and collaborators,Citation21 based on the ability of kinesins to walk along microtubules in the presence of ATP. In this assay, streptavidin is used to bridge biotinylated kinesins to the GUVs containing a small amount of biotinylated lipids (see Methods). In the presence of ATP, the kinesins bind to and walk along in vitro polymerized MTs attached to a coverslip, thereby pulling out nanotubes of average radius ~20 nm.
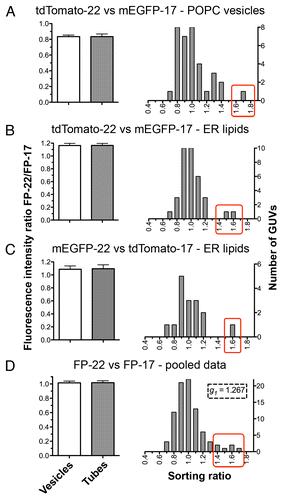
In order to compare the distribution of FP-17 and FP-22 in curved (tubes) and flat (vesicles) membranes, two single optical sections were acquired for each GUV, one in the equatorial plane of the vesicle and the other in the plane of the tube network (). The distribution of FP-22 and FP-17 was not homogenous within the tube network; in some tubes FP-22 was enriched compared with FP-17 (left side of the network, ), whereas in others the opposite was observed (right and upper side of the network, ). To analyze the global protein distribution within tube networks, we developed a manual quantification method described in Methods and shown in . We initially performed the analysis on the TA protein-containing GUVs of homogeneous (POPC) composition. This analysis did not reveal any statistically significant difference in the fluorescence intensity ratio of FP-22 over FP-17 in vesicles and tubes (, left panel). We then performed the same analysis using a physiological lipid mixture consisting of a total microsomal lipid extract. In this case too, no sorting between FP-22 and FP-17 was observed (, left panels). Although the average FP-22/FP-17 ratio was the same in tubes and vesicles, the distribution of the sorting ratio (defined as the fluorescence ratio of FP-22/FP-17 in tubes normalized by the same ratio in the corresponding vesicle) revealed in both POPC and microsomal lipid GUVs one or two outliers in which FP-22 was enriched in tubes compared with FP-17 (red rectangles, right graphs, ). When all the experimental data were pooled, skewness (g1) calculation confirmed the positive asymmetry of the distribution, suggesting a slight preference of FP-22 for curved membranes.
Figure 6. Quantitative analysis of the distribution of FP-17 and FP-22 in nanotubes and flat domains (GUVs). The graphs on the left show the averaged FP-22/FP-17 fluorescence intensity ratios in tube networks (gray bar) and in the corresponding GUVs (white bars). Results are given as mean ± SEM n = 35 (A and B), 16 (C), 86 (D). The histograms on the right show the distributions of sorting ratios (defined as the fluorescence ratio of FP-22/FP-17 in tubes normalized by the same ratio in the vesicle, see Methods) for the same data. The red boxes enclose outliers for which FP-22 was enriched in the tubular networks compared with FP-17.
We considered the possibility that the variability in FP-22 distribution could be due to a slow diffusion and consecutive failure to reach equilibrium during the time scale of our experiments. To address this issue, photobleaching was performed on tubes or vesicles in both the FP-17 and the FP-22 channels. As shown in , both FP-17 and FP-22 diffuse very rapidly within the vesicle with similar t1/2 (~15 s) and mobile fractions (89–90%). Similarly, no difference was detected between proteins grouped according to their fluorophore rather than according to their TMD, indicating that the fluorophore (tdTomato or mEGFP) has no effect on the diffusion of the TA proteins within the vesicle bilayer.
Figure 7. FRAP analysis of FP mobility in GUVs and in membrane tubes composed of ER lipids extracted from rat liver microsomes plus different FP-17/FP-22 combinations. (A–C) analysis in GUVs; D-F, analysis in nanotubes. (A, D) typical examples of FRAP experiments. The yellow rectangles indicate the bleached area. Scale bars, 5 μm. (B, E) Averaged fluorescence recovery curves for the indicated proteins. Bars represent S.E.M., n = 10 for each fluorescent protein. The red curves were obtained by fitting according to a mono-exponential equation (see Methods). (C, F) Average fluorescent half time of recovery (left) and mobile fractions (right) ± SEM ns, non significant, **, P = 0.0033 determined by Student’s t test.
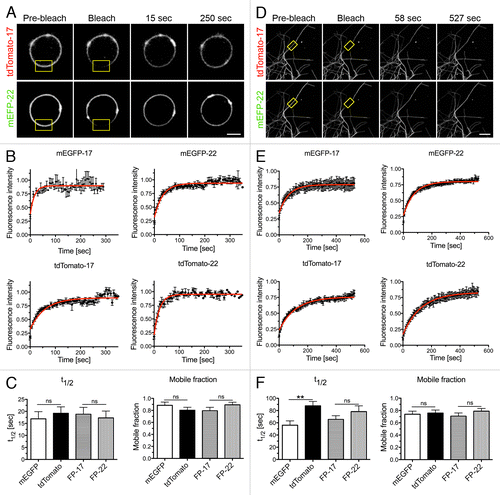
The same analysis, performed in the tubes (Fig. Seven D-F), also showed no statistically significant difference in the diffusion of the FP-22 and FP-17 constructs. However, recovery was slower for all four proteins (t1/2 ~1 min) and equilibrium was reached only after 8–10 min (four times longer than in vesicles), presumably because of the different geometry. These results suggest that exchange of FP-22 and -17 between tubular domains and between tubes and the vesicle occur at similar rate. Strikingly, however, we observed a difference in the diffusion rate of the two fluorescent variants within the tubes, regardless of TMD length. Both tdTomato-17 and -22 diffused more slowly than the corresponding mEGFP constructs, while the mobile fractions remained the same. A possible explanation for this observation is offered by the finding that ~50% of the proteins has inverted topology, with the N-terminal domain exposed to the lumen of the vesicle/tubule (). Since tdTomato is about twice as large as mEGFP, the narrow lumen of the tubules could slow diffusion of the population of protein with inverted topology.Citation26 However, since all the experiments were performed at least 20 min after adding kinesin and GUVs to the flow chamber to reach a steady-state, and since we performed reciprocal analyses (tdTomato-22 vs mEGFP-17 and mEGFP-22 vs tdTomato-17, see ), we conclude that the differences in the diffusion rates of the tdTomato and mEGFP constructs do not influence our analyses. In addition, the results of these experiments demonstrate that the mismatched FP-22 TMD does not drive the formation of large protein clusters, as predicted by Molecular Dynamics simulationsCitation16,Citation27 and experimentally verified in other systems.Citation28
Analysis of curvature-driven sorting in tubes pulled out by optical tweezers
We pursued our investigations in a more controlled system, in which the tube is pulled by optical tweezers from a GUV aspirated into a micropipette to set the membrane tension and precisely regulate the tube diameter ().Citation23 By measuring the bead displacement relative to the optical trap position with the progressive increase of the aspiration force (Fig. S2A), the variations of tube radius can be accurately calculated (see Methods). We applied the technique to measure the sorting of FP-17 and FP-22 with increasing membrane curvature (). displays the distribution of the FPs at the first tension step after the tube has been pulled out (upper panels, lowest tension and curvature) and at the final tension (lower panels, highest tension and curvature).
Figure 8. Distribution of TA proteins in GUVs and tubes of controlled curvature pulled from GUVs by optical tweezers and micropipette aspiration. (A) Illustration of the method. The GUV membrane is bound to a bead trapped by optical tweezers (right) and aspirated in a micropipette (left) to control its tension. By moving the GUV away from the trap, a tube can be pulled out between the GUV and the bead. Subsequently, by increasing the aspiration force, the membrane tension is progressively increased and the tube radius progressively decreased (see Methods). (B) Fluorescent confocal imaging of an aspirated GUV and membrane tube at initial (top) and final (bottom) tension. The increase in tension can be appreciated by the larger portion of the vesicle aspirated inside the micropipette and by the decreased fluorescent signal in the tube. In the lower row, the images have been nonlinearly enhanced to make the thinner tube visible. Scale bars in B and C, 5 μm. (C) Measurement of the sorting ratio of the indicated construct couples as a function of curvature (see Methods). The sorting ratio at the first tension step was set to 1. Results are given as means ± SEM (n = 17, 7, and 6 for FP-22/FP-17, mEGFP-17/tdTomato-17 and mEGFP-22/tdTomato-22 respectively). Red lines represent the linear regression fitting to the experimentally measured values.
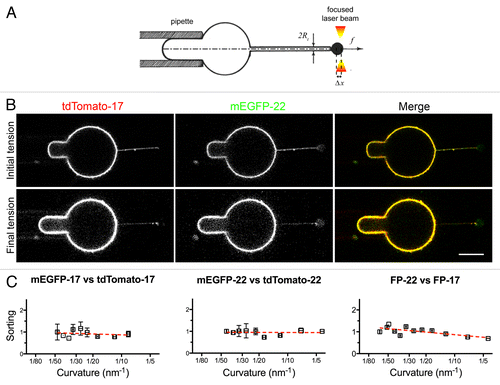
Quantitative analysis revealed that the sorting ratio of FP-22/FP-17 in tubes remained constant with increasing curvature (, right graph), like in control experiments performed with constructs bearing the same TMD but different fluorophores (, left and middle graphs). The analysis of the tube force f variations as a function of membrane tension σ and the linear relationship between f 2 and σ (Fig. S2B) also allows measuring, for the first time to our knowledge, the bending rigidity κ of bilayers made of microsomal lipids (9.4 kT ± 1.6, Fig. S2C). The values we obtained are consistent with the low cholesterol and sphingolipid content of ER membranesCitation29,Citation30 and with values obtained from synthetic bilayers mimicking the ER composition.Citation31,Citation32 Furthermore, the bending rigidity of the ER membranes is similar to that of GUVs composed of pure PC, the major lipid of the ER, and significantly lower than that of artificial membranes containing cholesterol/sphingomyelin 1:1 or cholesterol/DOPC 1:1 lipid mixtures.Citation5
Conclusions
Our previous work demonstrated a heterogeneous distribution of FP-22, distinct from that of FP-17, within the ER, at short times after its synthesis. 60–75 min after microinjection of FP-22 cDNA, the protein product could be detected at ERES and in ER tubules or at the rims of ER sheets but not within the sheets. On the basis of these observations, we deemed it unlikely that FP-22’s export from the ER is due to its interaction with a specific cargo receptor at ERES and hypothesized that the positively mismatched TMD partitions into ER domains (including ERES) to minimize the energy cost of the mismatched TMD-lipid bilayer assembly. Furthermore, since the common denominator between ERES, tubules and sheet rims is a high degree of membrane curvature, we postulated that membrane curvature might underlie the observed TMD-dependent sorting. Our reasoning was based on the knowledge that tilting of the TMD within the bilayer is a common way of accommodating positive mismatch,Citation33,Citation34 but that this arrangement results in suboptimal interaction between the TMD and the acyl chains (which are roughly perpendicular to the plane of the membrane). We speculated that in curved domains, because of a more irregular disposition of the phospholipid molecules and increased splaying of the acyl chains in the inner monolayer, there might be a better fit between a tilted TMD and the surrounding bilayer. The present study was designed to test this hypothesis in the simplified setting of membrane tubes mechanically pulled from GUVs.
Using two different methods to generate nanotubes from GUVs of either homogeneous lipid composition (POPC) or made of ER lipids, we failed to detect any clear preferential partitioning of FP-22 into nanotubes. Thus our study, in addition to characterizing the orientation of transmembrane proteins in reconstituted GUVs and to determining the bending rigidity of bilayers composed of microsomal lipids and TA proteins, demonstrates that curvature alone cannot induce TMD-dependent sorting of single-pass polypeptides in the ER environment. These results strongly suggest that proteins are involved in TMD-dependent sorting at the ER-Golgi interface. It is interesting that a very recent study with GUVs composed of lipid mixtures somewhat different from ours has instead demonstrated curvature-dependent sorting of a polytopic protein, the bacterial potassium channel KvAP.Citation35
There are various ways in which proteins could play a role in TMD-dependent partitioning and subsequent export of FP-22. For instance, FP-22’s mismatched TMD could interact with low affinity with a number of membrane proteins enriched at ERES (cargo receptors or cargo molecules themselvesCitation36 and in tubular ER (e.g., reticulons and DP1 proteinsCitation4). Alternatively, proteins could stabilize lipid subdomains, into which FP-22 would preferentially partition. Although ER lipids themselves do not spontaneously undergo lateral phase separation, there is evidence for raft-like domains in the ERCitation37-Citation39 and for lipid compositional differences in COPII transport vesicles compared with the bulk ER.Citation40-Citation42 The formation of these domains is presumably aided by proteins. In model systems, it has been demonstrated that curvature-induced lipid sorting is efficient only when the membrane composition is close to phase separation and is aided by lipid-binding proteins.Citation6 Moreover, the presence of transmembrane proteins can induce lateral phase separation in the lipid bilayer.Citation43-Citation45 A role for lipid domains in TMD-dependent sorting in the ER is further suggested by our previous investigation on proteoliposomes containing TA proteins of differing TMD length, in which differential scanning calorimetry and fluorescent measurements of lipid probes were applied to analyze partitioning.Citation46 Further studies in model systems as well as in cells will be required to establish how a combination of lipids, proteins and membrane geometry results in TMD-dependent partitioning and sorting at the ER-Golgi interface.
Materials and Methods
Plasmid construction
The mEGFP-17 and 22 constructs under the CMV promoter () have been described previously.Citation20 The equivalent constructs, containing the tdTomato fluorescent protein,Citation25 were obtained by amplifying the tdTomato sequence from ptdTomato-N1 (from Clontech) by PCR using a reverse primer designed with a 3′ end containing a BspEI recognition site. The amplified product was digested with NheI and BspEI at the 5′ and 3′ ends, respectively, and then ligated into pmCerulean-17 or -22Citation12 after excising the mCerulean sequence with the same two restriction enzymes.
To produce recombinant proteins, the FP sequences were subcloned into the pGEX2T vector (). mEGFP-17/22 and tdTomato 17/22 were amplified using forward primers designed to introduce an EcoRI site upstream to the open reading frame; the amplified fragments were digested with EcoRI at both the 5′ and 3′ ends and ligated into the EcoR1- digested pGEX2T vector (GE Healthcare). All recombinant plasmids were checked by sequencing.
Cell culture, transfection, and imaging of transfected cells
CV1 cells were grown on coverslips in DMEM supplemented with 10% Fetal Bovine Serum and antibiotics as previously described.Citation20 Cells were transfected with the Calcium Phosphate method and fixed with paraformaldehyde 24 h after transfection. The coverslips were mounted in Mowiol and observed with a Zeiss LSM 510 Meta confocal system (Carl Zeiss, Oberkochen, Germany) using a 63x PlanApo lens. EGFP was imaged with the use of the 488 nm line of an Argon laser, a 405/488/543/633 dichroic mirror and a 505–550 band pass emission filter. tdTomato was excited at 543 nm with a He-Ne laser and imaged using the same dichroic and a 560 long-pass emission filter.
Preparation of recombinant proteins
FP-17 and 22 recombinant proteins, in 30 mM N-octyl glucoside (OG) detergent, were prepared essentially as described for cytochrome b5 constructs,Citation46 except that incubation of the soluble portion of the bacterial lysate with GST beads (GS4B from GE Healthcare) and digestion of the attached fusion protein with thrombin were shortened to 1 h and 2 h respectively. Protein preparations were checked by SDS-PAGE followed by Coomassie Blue staining and quantified with the BCA assay (Pierce/Thermo Fisher Scientific, Waltham, MA). The final recombinant protein concentration, estimated with the BCA assay, was corrected for the presence on the gel of minor bands (quantified with the Image J software) corresponding to degradation products of the recombinant proteins.
Extraction of microsomal lipids
Rat liver microsomes were isolated by differential centrifugation.Citation47 Microsomes were resuspended in 250 mM sucrose, 5 mM TRIS-HCl pH 7.4, 0.1 mM EDTA, divided into aliquots, frozen in liquid nitrogen and stored at -80 °C. The protein content was measured with the BCA assay.
500 μl of ER suspension (containing ~10 mg protein) were extracted with organic solvents according to.Citation48 The organic phase was dried with a Rotavapor and the lipid film resuspended in 500 μl CHCl3 and stored at -20 °C. Phospholipid concentration was determined by the Ames procedure.Citation49
Preparation of Large Unilamellar Vesicles (LUVs)
600 μg of POPC (from Avanti Polar Lipids) or ER lipids extracted from rat liver microsomes in chloroform were dried with a Rotavapor for 1 h and then resuspended overnight in 400 mM KCl, 4 mM TRIS-HCl pH 7.4 and 30 mM OG. A small amount of biotinylated lipids (from AvantiPolar Lipids), required to pull nanotubes from GUVs (see below), were included in the reconstitution sample: 1% mol/mol Biotinyl-Cap-dioleyl- phosphatidylethanolamine for experiments with kinesin motors or 0.03% w/w distearoyl-phosphatidylethanolamine- Peg(2000)- Biotin for experiments with the optical tweezers. Different combinations of recombinant FP-17 and FP-22 were added to the detergent-lipid mix at a molar protein/lipid ratio of 1:500. The samples were than dialysed (12.000–14.000 Mr Cutoff) for 4 d against 8 x 2 L of 400 mM KCl, 4 mM TRIS-HCl pH 7.4.
Electron microscopy of negatively stained proteoliposomes
Formvar carbon-coated nickel grids were placed on top of a 30 μl drop of proteoliposome suspension for 10 min on ice. Grids were then washed quickly 5 times in H2O, stained for 10 min on ice with 1% uranyl acetate. After draining the excess uranyl acetate on a Whatman filter, grids were observed under a Philips CM10 transmission electron microscope.
Carbonate extraction and alkaline sucrose gradient centrifugation
Proteoliposome suspensions (or recombinant proteins without lipids) were treated with an equal volume of 0.2 M Na2CO3 for 30 min on ice and then brought to 1.2 M sucrose in 0.1 M Na2CO3 in a final volume of 740 μl. The samples were layered under discontinuous sucrose gradients composed of five layers (560 μl each of 1, 0.5, 0.25, 0.15 and 0 M sucrose in 0.1 M Na2CO3). After ultracentrifugation overnight at 40000 rpm at 4 °C (Beckman SW 55 rotor), fractions of 0.7 ml each were collected, precipitated with 2.7 ml of 20% TCA, washed with acetone, resuspended in PBS and analyzed by SDS-PAGE- Western Blot. Polyclonal anti-GFP (Abcam) and peroxidase-conjugated anti-rabbit IgG (Jackson Immunoresearch Laboratories) were used as primary and secondary immunostaining reagents, respectively.
Electroformation of Giant Unilamellar Vesicles (GUVs)
The proteoliposomes generated by detergent dialysis were used to generate GUVs by electroformation (reviewed in ref. Citation24). The proteoliposomes were first re-equilibrated in 100 mM KCl, using Midi-Trap G25 desalting columns (GE Healthcare), then pelleted by ultracentrifugation at 55000 rpm for 2 h at 4 °C (Beckman TLS 55 rotor), and finally resuspended in an appropriate volume of 100 mM KCl, 4 mM TRIS-HCl pH 7.4 to obtain a final lipid concentration of 1.3 mg/ml. 1, 1.5 or 2 μl drops of LUV suspension were placed on the conductive side of two ITO-coated slides (Präzisions Glas and Optik GmbH) and dried overnight under vacuum. The electroformation chamber was built with two liposome-coated ITO slides with their conductive sides facing each other, separated by a 1 mm Teflon spacer, connected to a low frequency generator (TG315 function generator, TTi Thurlby Thandar Instruments) via adhesive copper electrodes and sealed with Sigilum wax (Vitrex Medical A/S). The dried proteoliposomes were rehydrated by filling the chamber (of about 1.5 ml) with a sucrose solution of controlled osmotic pressure (180 mOsm or 279 mOsm for experiments with kinesin motors or with the optical tweezers respectively). A sinusoidal electric field with an increasing voltage ramp from 20 mV to 1.1 V (6 min duration for each voltage step) at 10 Hz frequency was applied for 3–4 h. After satisfactory GUV growth, checked by phase contrast (Axiovert 200M, Carl Zeiss – Fig. S1), 20 μl of vesicles were taken from the center of the area of initial liposome deposition.
Kinesin tube assay
Membrane tube pulling by kinesin motors from GUVs was performed essentially as described.Citation21,Citation24 Briefly, in vitro polymerized microtubules were allowed to adhere to a coverslip, which had been sealed to a slide with melted parafilm to constitute a flow chamber. Biotinylated and truncated kinesin-1 (KinBio401 from D. melanogaster) motors (a gift of F. Nédélec, European Molecular Biology Laboratory, Heidelberg) were purified from E. coli expressing the kinesin-BCCP-H6 plasmid (Addgene, Plasmid 15960: pWC2), using a standard protocol for His-tagged proteins.Citation50
Ten μl of a preassembled complex of biotinylated kinesin and streptavidin (Sigma Aldrich) in IMI buffer (50 mM Imidazole pH 6.7, 50 mM NaCl, 2 mM EGTA, 1 mM MgCl2) supplemented with 5 mg/ml casein and 30 µM taxol were introduced into the chamber. After a 15 min incubation at room temperature, 10 μl of motility buffer (IMI buffer supplemented with 30 µM taxol, 1 mM ATP, 5 mM DTT, 25 mM glucose, 0.18 mg/ml catalase and 0.37 mg/ml glucose oxidase) and 1 μl of GUVs were loaded into the chamber, which was sealed with Sigilum wax on both sides and incubated for 15 min at room temperature to allow kinesins to pull nanotubes.
To measure the distribution of the FPs in tubes and in vesicles, single optical sections were acquired for both FP-17 and FP-22 channels in the vesicle plane and in the tube plane using a Nikon A1R confocal microscope equipped with a Plan apo 63x 1.3 NA objective. mEGFP and tdTomato were excited at 488 nm and 561 nm respectively. Acquisition settings were adjusted to avoid pixel saturation.
Optical tweezers tube assay
The pulling of tubes of defined diameter from GUVs by a combination of optical tweezers and micropipette aspiration was performed as described,Citation5,Citation24 see ). Pipettes were prepared from borosilicate glass capillaries (internal radius 0.7 mm, external radius 1 mm, from Kimble Glass Inc.), using a pipette puller (Sutter instrument P-2000). The pipette radius was set to ~4 μm using a microforge microscope (MF-800 Narishige, Japan). After inserting the micropipette (filled with 10 mg/ml casein) into a flow chamber filled with 200 μl of HKM buffer (50 mM Hepes pH 7.2, 120 mM KAcetate, 1 mM MgCl2 and 2 mM EGTA), 2–3 μl of streptavidin-coated polystyrene beads (3 μm in diameter, from Spherotech Inc.) followed by of 5–10 μl of GUVs were injected into the chamber. After selecting an optically fluctuating vesicle, the zero reference pressure was set by adjusting the level of water in a reservoir connected to the micropipette, and the vesicle was aspirated by slightly increasing the suction pressure. A bead was then trapped by the optical tweezers created by a focused infrared laser beam (Ytterbium fiber laser 1070 nm, 5W, IPG GmBH) and contacted to the biotinylated lipid-containing GUV. A single tube was pulled by moving the GUV away from the trap. The aspiration force was then progressively increased step by step, where one step typically corresponds to a vertical movement of the water reservoir of 0.5 mm. For each tension step, one equatorial confocal plane, in which both the vesicle and tube were in focus, was acquired with the same laser settings as used for the kinesin assay. A DIC image was also acquired at each step
The force f acting on the optically trapped bead was measured by tracking the position of the bead x relative to the trap center x0 according to f = -k (x - x0), where k is the trap stiffness. Calibration of the optical trap by the viscous drag method yielded a trap stiffness of k = 200 ± 10 pN µm−1 W−1. Single particle tracking of the bead imaged by transmission was performed using a Matlab program (kindly provided by Gil Toombes and Patricia Bassereau, UMR168, CNRS-Institut Curie, Paris). The GUV membrane tension σ was deduced from Laplace’s lawCitation51 σ = ΔP × Rpip /(2(1-Rpip/Rves)) where Rpip is the pipette radius, Rves is the vesicle radius, and ΔP is the aspiration pressure given by the vertical displacement of the water reservoir linked to the micropipette. From the tube force f and the membrane tension σ, we deduced the radius of the tube Rtube = f/4πσ and its curvature c = 1/Rtube. The bending rigidity of the microsomal lipid bilayer was derived from the linear relationship between the squared force f2 applied on the bead and the membrane tension σ, according to the following equation: f2 = (8 π2 κ) σ.
Treatment of GUVs with PK
We used two experimental set-ups. The first set-up was used to look at the steady-state effect of PK addition. In the same chambers as described for the membrane tube pulling assay by kinesin motors, we incubated 2 µL GUVs for at least 10 min in either a control buffer (50 mM Hepes, pH 7.2, 120 mM potassium acetate, 1mM MgCl2) or in the same buffer containing PK (4–20 µg/mL). In the second set-up, we used open chambers made of a hollow rectangular prism of polydimethylsiloxane in which a small (5 mm diameter) hole was cut, to directly inject the PK solution into the GUV suspension (25 µL) and initiate visualization immediately thereafter. Although the flow created by PK injection displaced the GUVs within the chamber and prevented the visualization of the same GUVs before and after PK addition, the time taken for the PK to diffuse in the chamber allowed us to follow the effect of PK addition by time-lapse confocal video-microscopy. Triton X100 was used at low concentration (0.01%) to permeabilize the GUVs after PK injection.
Image quantification
To analyze the distribution of FP-17 and FP-22 in tube networks pulled by kinesin motors, we used a manual method in Image J (illustrated in ). First, the background maximum intensity was measured and subtracted from the image. Then, a region of interest (ROI) was drawn around the tube network or the vesicle. A manual threshold was then applied to create a mask highlighting only the tube network or the vesicle. This mask was superimposed on the original image (with the image calculator tool and the AND operator) and the mean fluorescence intensity was measured in both red and green channels.
To analyze the distribution of FP-17 and FP-22 in tubes pulled by optical tweezers, we calculated the sorting ratio defined as the fluorescence intensity ratio of FP-22 over FP-17 in the tube normalized to the same ratio in the vesicle, using a Matlab routine kindly provided by Sophie Aimon and Patricia Bassereau (UMR168, CNRS-Institut Curie, Paris).Citation6 Briefly, a rectangular selection was drawn around the horizontal tube and an averaged fluorescence profile along the axis perpendicular to the tube was generated. The same operation was applied to a selected portion of the vesicle. The sorting ratios were averaged and plotted against membrane curvature c = 1/Rtube.Citation51
Fluorescence Recovery after Photobleaching (FRAP)
For FRAP experiments on vesicles and tubular networks pulled out by kinesin motors, three pre-bleached images were acquired, and a portion of the vesicle or of the tube (3 x 4 μm or 3 x 9 μm, respectively) was bleached (4–6 iterations at 100% laser power). Recovery was then followed by acquiring images every 6 s. After background subtraction (determined in an area outside the GUV), fluorescence in the bleached area was determined using the ImageJ software, and normalized to the total vesicle or tubular network fluorescence. The half recovery time t1/2 and the mobile fraction were derived from experimentally measured data by fitting the following monoexponential equation giving the fluorescence F as a function of time t: F(t) = Fpost + (Frec – Fpost)(1-exp(-t/τ)), where Fpost is the fluorescence signal after photobleaching, Frec is the maximum fluorescence recovery value and τ is the time constant.
Significance tests
Statistical significance between the distribution of FP-22 and 17 in tubes and in vesicles was evaluated by paired Student’s t test. The unpaired t test was used to evaluate the statistical significance of the difference between fluorescence levels of GUVs treated or not treated with PK. Differences between the t1/2 and the mobile fraction of the two proteins in FRAP experiments were evaluated by unpaired Student’s t test. In both cases, the significance level was set to 5%. Skewness calculation of frequency distributions (g1) was performed using Prism Software. Values are given as averages ± SEM (standard error mean).
| Abbreviations: | ||
| ER | = | endoplasmic reticulum |
| ERES | = | ER exit sites |
| FP | = | fluorescent protein |
| FRAP | = | Fluorescence recovery after photobleaching |
| GUV | = | giant unilamellar vesicle |
| LUV | = | large unilamellar vesicle |
| OG | = | N-octyl glucoside |
| PK | = | Protease K |
| POPC | = | palmitoyl oleyl phosphatidylcholine |
| TA | = | tail-anchored |
| TMD | = | transmembrane domain |
Additional material
Download Zip (2.4 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Work performed in the laboratory of NB was supported by a grant from the Italian Cancer Association (AIRC, Investigator Grant 2009). MF was a doctoral student of the University of Milan and recipient of a short-term EMBO fellowship for collaborative work at the Institut Curie. We thank Sara Colombo for helpful discussions and support and the Fondazione Monzino (Milan) for its generous gift of the Zeiss LSM 510 Meta confocal microscope. We thank Patricia Bassereau for technical support and discussion, David Guet and Mathieu Pinot for assistance with the optical tweezers set-up and funding from Institut Curie, CNRS (Program ‘Prise de risque à l’interface Physique-Biologie’), ANR (grant number ANR09-JCJC-0020–01).
Supplementary Material
Supplementary material may be downloaded here: www.landesbioscience.com/journals/cellularlogistics/article/29087/
References
- McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005; 438:590 - 6; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16319878 http://dx.doi.org/10.1038/nature04396; PMID: 16319878
- Johannes L, Mayor S. Induced domain formation in endocytic invagination, lipid sorting, and scission. Cell 2010; 142:507 - 10; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20723749 http://dx.doi.org/10.1016/j.cell.2010.08.007; PMID: 20723749
- Stachowiak JC, Brodsky FM, Miller EA. A cost-benefit analysis of the physical mechanisms of membrane curvature. Nat Cell Biol 2013; 15:1019 - 27; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23999615 http://dx.doi.org/10.1038/ncb2832; PMID: 23999615
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006; 124:573 - 86; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16469703 http://dx.doi.org/10.1016/j.cell.2005.11.047; PMID: 16469703
- Roux A, Cuvelier D, Nassoy P, Prost J, Bassereau P, Goud B. Role of curvature and phase transition in lipid sorting and fission of membrane tubules. EMBO J 2005; 24:1537 - 45; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15791208 http://dx.doi.org/10.1038/sj.emboj.7600631; PMID: 15791208
- Sorre B, Callan-Jones A, Manneville JB, Nassoy P, Joanny JF, Prost J, Goud B, Bassereau P. Curvature-driven lipid sorting needs proximity to a demixing point and is aided by proteins. Proc Natl Acad Sci U S A 2009; 106:5622 - 6; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19304798 http://dx.doi.org/10.1073/pnas.0811243106; PMID: 19304798
- Vanni S, Vamparys L, Gautier R, Drin G, Etchebest C, Fuchs PF, Antonny B. Amphipathic lipid packing sensor motifs: probing bilayer defects with hydrophobic residues. Biophys J 2013; 104:575 - 84; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23442908 http://dx.doi.org/10.1016/j.bpj.2012.11.3837; PMID: 23442908
- Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 2003; 114:497 - 509; http://dx.doi.org/10.1016/S0092-8674(03)00609-3; PMID: 12941277
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell 2004; 116:153 - 66; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14744428 http://dx.doi.org/10.1016/S0092-8674(03)01079-1; PMID: 14744428
- Sharpe HJ, Stevens TJ, Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010; 142:158 - 69; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20603021 http://dx.doi.org/10.1016/j.cell.2010.05.037; PMID: 20603021
- Karsten V, Hegde RS, Sinai AP, Yang M, Joiner KA. Transmembrane domain modulates sorting of membrane proteins in Toxoplasma gondii.. J Biol Chem 2004; 279:26052 - 7; http://dx.doi.org/10.1074/jbc.M400480200; PMID: 15056659
- Ronchi P, Colombo S, Francolini M, Borgese N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J Cell Biol 2008; 181:105 - 18; http://dx.doi.org/10.1083/jcb.200710093; PMID: 18391072
- Dukhovny A, Yaffe Y, Shepshelovitch J, Hirschberg K. The length of cargo-protein transmembrane segments drives secretory transport by facilitating cargo concentration in export domains. J Cell Sci 2009; 122:1759 - 67; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19435807 http://dx.doi.org/10.1242/jcs.039339; PMID: 19435807
- Mercanti V, Marchetti A, Lelong E, Perez F, Orci L, Cosson P. Transmembrane domains control exclusion of membrane proteins from clathrin-coated pits. J Cell Sci 2010; 123:3329 - 35; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20826467 http://dx.doi.org/10.1242/jcs.073031; PMID: 20826467
- Munro S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J 1995; 14:4695 - 704; http://www.ncbi.nlm.nih.gov/pubmed/7588599 PMID: 7588599
- Schmidt U, Weiss M. Hydrophobic mismatch-induced clustering as a primer for protein sorting in the secretory pathway. Biophys Chem 2010; 151:34 - 8; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20537786 http://dx.doi.org/10.1016/j.bpc.2010.04.009; PMID: 20537786
- Sato K, Sato M, Nakano A. Rer1p, a retrieval receptor for ER membrane proteins, recognizes transmembrane domains in multiple modes. Mol Biol Cell 2003; 14:3605 - 16; http://dx.doi.org/10.1091/mbc.E02-12-0777; PMID: 12972550
- Herzig Y, Sharpe HJ, Elbaz Y, Munro S, Schuldiner M. A systematic approach to pair secretory cargo receptors with their cargo suggests a mechanism for cargo selection by Erv14. PLoS Biol 2012; 10:e1001329; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22629230 http://dx.doi.org/10.1371/journal.pbio.1001329; PMID: 22629230
- Borgese N, Brambillasca S, Colombo S. How tails guide tail-anchored proteins to their destinations. Curr Opin Cell Biol 2007; 19:368 - 75; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17629691 http://dx.doi.org/10.1016/j.ceb.2007.04.019; PMID: 17629691
- Bulbarelli A, Sprocati T, Barberi M, Pedrazzini E, Borgese N. Trafficking of tail-anchored proteins: transport from the endoplasmic reticulum to the plasma membrane and sorting between surface domains in polarised epithelial cells. J Cell Sci 2002; 115:1689 - 702; PMID: 11950887
- Roux A, Cappello G, Cartaud J, Prost J, Goud B, Bassereau P. A minimal system allowing tubulation with molecular motors pulling on giant liposomes. Proc Natl Acad Sci U S A 2002; 99:5394 - 9; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11959994 http://dx.doi.org/10.1073/pnas.082107299; PMID: 11959994
- Leduc C, Campàs O, Zeldovich KB, Roux A, Jolimaitre P, Bourel-Bonnet L, Goud B, Joanny JF, Bassereau P, Prost J. Cooperative extraction of membrane nanotubes by molecular motors. Proc Natl Acad Sci U S A 2004; 101:17096 - 101; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15569933 http://dx.doi.org/10.1073/pnas.0406598101; PMID: 15569933
- Callan-Jones A, Sorre B, Bassereau P. Curvature-driven lipid sorting in biomembranes. Cold Spring Harb Perspect Biol 2011; 3:•••; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21421916 http://dx.doi.org/10.1101/cshperspect.a004648; PMID: 21421916
- Manneville JB, Leduc C, Sorre B, Drin G. Studying in vitro membrane curvature recognition by proteins and its role in vesicular trafficking. Methods Cell Biol 2012; 108:47 - 71; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22325597 http://dx.doi.org/10.1016/B978-0-12-386487-1.00003-1; PMID: 22325597
- Nienhaus GU, Wiedenmann J. Structure, dynamics and optical properties of fluorescent proteins: perspectives for marker development. Chemphyschem 2009; 10:1369 - 79; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19229892 http://dx.doi.org/10.1002/cphc.200800839; PMID: 19229892
- Domanov YA, Aimon S, Toombes GE, Renner M, Quemeneur F, Triller A, Turner MS, Bassereau P. Mobility in geometrically confined membranes. Proc Natl Acad Sci U S A 2011; 108:12605 - 10; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21768336 http://dx.doi.org/10.1073/pnas.1102646108; PMID: 21768336
- Parton DL, Klingelhoefer JW, Sansom MS. Aggregation of model membrane proteins, modulated by hydrophobic mismatch, membrane curvature, and protein class. Biophys J 2011; 101:691 - 9; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21806937 http://dx.doi.org/10.1016/j.bpj.2011.06.048; PMID: 21806937
- Botelho AV, Huber T, Sakmar TP, Brown MF. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys J 2006; 91:4464 - 77; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17012328 http://dx.doi.org/10.1529/biophysj.106.082776; PMID: 17012328
- Sprong H, van der Sluijs P, van Meer G. How proteins move lipids and lipids move proteins. Nat Rev Mol Cell Biol 2001; 2:504 - 13; http://dx.doi.org/10.1038/35080071; PMID: 11433364
- van Meer G, Sprong H. Membrane lipids and vesicular traffic. Curr Opin Cell Biol 2004; 16:373 - 8; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15261669 http://dx.doi.org/10.1016/j.ceb.2004.06.004; PMID: 15261669
- Loftus AF, Hsieh VL, Parthasarathy R. Modulation of membrane rigidity by the human vesicle trafficking proteins Sar1A and Sar1B. Biochem Biophys Res Commun 2012; 426:585 - 9; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22974979 http://dx.doi.org/10.1016/j.bbrc.2012.08.131; PMID: 22974979
- Settles EI, Loftus AF, McKeown AN, Parthasarathy R. The vesicle trafficking protein Sar1 lowers lipid membrane rigidity. Biophys J 2010; 99:1539 - 45; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20816066 http://dx.doi.org/10.1016/j.bpj.2010.06.059; PMID: 20816066
- de Planque MR, Killian JA. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol Membr Biol 2003; 20:271 - 84; http://dx.doi.org/10.1080/09687680310001605352; PMID: 14578043
- Kim T, Im W. Revisiting hydrophobic mismatch with free energy simulation studies of transmembrane helix tilt and rotation. Biophys J 2010; 99:175 - 83; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20655845 http://dx.doi.org/10.1016/j.bpj.2010.04.015; PMID: 20655845
- Aimon S, Callan-Jones A, Berthaud A, Pinot M, Toombes GE, Bassereau P. Membrane shape modulates transmembrane protein distribution. Dev Cell 2014; 28:212 - 8; http://www.ncbi.nlm.nih.gov/pubmed/24480645 http://dx.doi.org/10.1016/j.devcel.2013.12.012; PMID: 24480645
- D'Arcangelo JG, Stahmer KR, Miller EA. Vesicle-mediated export from the ER: COPII coat function and regulation. Biochim Biophys Acta 2013; 1833: 2464-72; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23419775
- Browman DT, Resek ME, Zajchowski LD, Robbins SM. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci 2006; 119:3149 - 60; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16835267 http://dx.doi.org/10.1242/jcs.03060; PMID: 16835267
- Campana V, Sarnataro D, Fasano C, Casanova P, Paladino S, Zurzolo C. Detergent-resistant membrane domains but not the proteasome are involved in the misfolding of a PrP mutant retained in the endoplasmic reticulum. J Cell Sci 2006; 119:433 - 42; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16443748 http://dx.doi.org/10.1242/jcs.02768; PMID: 16443748
- Smith DC, Sillence DJ, Falguières T, Jarvis RM, Johannes L, Lord JM, Platt FM, Roberts LM. The association of Shiga-like toxin with detergent-resistant membranes is modulated by glucosylceramide and is an essential requirement in the endoplasmic reticulum for a cytotoxic effect. Mol Biol Cell 2006; 17:1375 - 87; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16381816 http://dx.doi.org/10.1091/mbc.E05-11-1035; PMID: 16381816
- Runz H, Miura K, Weiss M, Pepperkok R. Sterols regulate ER-export dynamics of secretory cargo protein ts-O45-G. EMBO J 2006; 25:2953 - 65; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16794576 http://dx.doi.org/10.1038/sj.emboj.7601205; PMID: 16794576
- Sturbois-Balcerzak B, Vincent P, Maneta-Peyret L, Duvert M, Satiat-Jeunemaitre B, Cassagne C, Moreau P. ATP-Dependent formation of phosphatidylserine-rich vesicles from the endoplasmic reticulum of leek cells. Plant Physiol 1999; 120:245 - 56; http://dx.doi.org/10.1104/pp.120.1.245; PMID: 10318702
- Bonnon C, Wendeler MW, Paccaud JP, Hauri HP. Selective export of human GPI-anchored proteins from the endoplasmic reticulum. J Cell Sci 2010; 123:1705 - 15; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20427317 http://dx.doi.org/10.1242/jcs.062950; PMID: 20427317
- Nielsen SO, Lopez CF, Ivanov I, Moore PB, Shelley JC, Klein ML. Transmembrane peptide-induced lipid sorting and mechanism of Lalpha-to-inverted phase transition using coarse-grain molecular dynamics. Biophys J 2004; 87:2107 - 15; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15454415 http://dx.doi.org/10.1529/biophysj.104.040311; PMID: 15454415
- Domanski J, Marrink SJ, Schafer LV. Transmembrane helices can induce domain formation in crowded model membranes. Biochim Biophys Acta 2012; 1818: 984-94; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21884678
- Yin F, Kindt JT. Atomistic simulation of hydrophobic matching effects on lipid composition near a helical peptide embedded in mixed-lipid bilayers. J Phys Chem B 2010; 114:8076 - 80; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20509701 http://dx.doi.org/10.1021/jp100931h; PMID: 20509701
- Ceppi P, Colombo S, Francolini M, Raimondo F, Borgese N, Masserini M. Two tail-anchored protein variants, differing in transmembrane domain length and intracellular sorting, interact differently with lipids. Proc Natl Acad Sci U S A 2005; 102:16269 - 74; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16258060 http://dx.doi.org/10.1073/pnas.0508157102; PMID: 16258060
- Walter P, Blobel G. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol 1983; 96:84 - 93; http://dx.doi.org/10.1016/S0076-6879(83)96010-X; PMID: 6656655
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957; 226:497 - 509; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=13428781 PMID: 13428781
- Ames BN, Dubin DT. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J Biol Chem 1960; 235:769 - 75; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=13793161 PMID: 13793161
- Surrey T, Elowitz MB, Wolf PE, Yang F, Nédélec F, Shokat K, Leibler S. Chromophore-assisted light inactivation and self-organization of microtubules and motors. Proc Natl Acad Sci U S A 1998; 95:4293 - 8; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9539730 http://dx.doi.org/10.1073/pnas.95.8.4293; PMID: 9539730
- Kwok R, Evans E. Thermoelasticity of large lecithin bilayer vesicles. Biophys J 1981; 35:637 - 52; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7272454 http://dx.doi.org/10.1016/S0006-3495(81)84817-5; PMID: 7272454