
Abstract
Genetic risk factors for chronic kidney disease (CKD) are being identified through international collaborations. By comparison, epigenetic risk factors for CKD have only recently been considered using population-based approaches. DNA methylation is a major epigenetic modification that is associated with complex diseases, so we investigated methylome-wide loci for association with CKD. A total of 485,577 unique features were evaluated in 255 individuals with CKD (cases) and 152 individuals without evidence of renal disease (controls). Following stringent quality control, raw data were quantile normalized and β values calculated to reflect the methylation status at each site. The difference in methylation status was evaluated between cases and controls with resultant P values adjusted for multiple testing. Genes with significantly increased and decreased levels of DNA methylation were considered for biological relevance by functional enrichment analysis using KEGG pathways in Partek Genomics Suite. Twenty-three genes, where more than one CpG per loci was identified with Padjusted < 10−8, demonstrated significant methylation changes associated with CKD and additional support for these associated loci was sought from published literature. Strong biological candidates for CKD that showed statistically significant differential methylation include CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2 genes; several genes are differentially methylated in kidney tissue and RNA-seq supports a functional role for differential methylation in ELMO1 and PRKAG2 genes. This study reports the largest, most comprehensive, genome-wide quantitative evaluation of DNA methylation for association with CKD. Evidence confirming methylation sites influence development of CKD would stimulate research to identify epigenetic therapies that might be clinically useful for CKD.
Introduction
Chronic kidney disease (CKD) with multifactorial etiology leading to end-stage renal disease (ESRD) is a significant public health concern with increasing numbers of individuals affected worldwide.Citation1-Citation3 CKD is defined as an estimated glomerular filtration rate (eGFR) <60 mL/min/1.73m2.Citation4 ESRD (also known as stage 5 CKD), is defined as an eGFR <15 mL/min/1.73m2, and is the most severe form of CKD; many individuals with ESRD require renal replacement therapy, such as dialysis or kidney transplantation. In 2010, 50 965 adult UK patients received renal replacement therapy reflecting a UK ESRD prevalence of 832 per million population and a 63% increase in renal replacement therapy population over the past decade.Citation5 Currently, more than half of UK healthcare costs for CKD are spent on renal replacement therapy.Citation6 CKD leads to reduced quality of life for affected individuals and is associated with an increased risk of premature death.Citation7
There is an urgent need to identify biological markers that help identify individuals who are at higher risk of developing CKD so that targeted therapies can be used to delay the onset or progression of CKD. An inherited predisposition to CKD exists, and research to date has focused on common genetic variants in large cohorts of individuals.Citation8-Citation10 Nevertheless, currently identified genetic markers account for a limited component of the heritability for this disorder. More integrated strategies that explore the interaction between an individual’s genome and environmental stimuli are required to determine inherited risks for CKD.Citation11-Citation13 It is hypothesized that the genetic background interacts with multiple potential modifying stimuli including intrauterine environment, nutrition, toxins, pathogens, and drugs, to influence the risk of development and progression of CKD.Citation14 While it is possible that rare genetic variants, copy number variation, or mitochondrial mutations account for some of the “missing heritability,” it is also plausible that epigenetic phenomena may be critical determinants of CKD.Citation15,Citation16
Emerging evidence for epigenetic phenomena has revolutionized investigations of heritable influences on disease and, complementary to GWAS, it is now cost-effective to perform population-based studies of the epigenome (EWAS).Citation17-Citation19 Epigenetic changes have been associated with CKDCitation15,Citation16,Citation20-Citation22 and the development of novel genetic-epigenetic risk profiles are key translational medicine priorities for targeted therapies ().Citation15,Citation23,Citation24 Drugs that directly target the epigenome are already in clinical use for the treatment of cancer and, potentially, such drugs may have efficacy in the treatment of CKD.Citation25
Figure 1. The impact of epigenetics for chronic kidney disease. A role for epigenetics for chronic kidney disease.
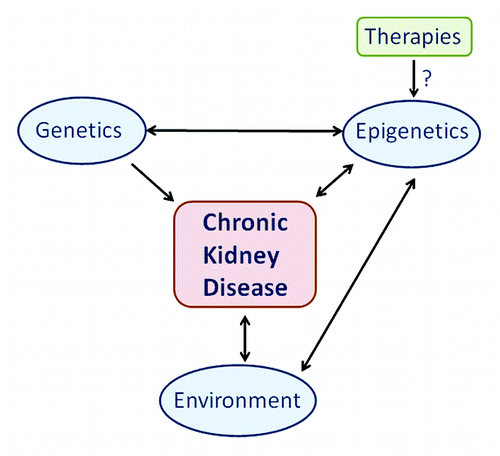
Methylation is a key epigenetic feature of DNA that plays an important role in chromosomal integrity and the regulation of gene expression with different methylation profiles now being associated with many complex diseases.Citation26 DNA methyltransferases are required for the process of DNA methylation and sequence variants in these genes have been associated with diabetic kidney disease.Citation27 Preliminary research evidence provides support for the importance of differential methylation in CKD.Citation28-Citation30 We describe a relatively large-scale case-control study that aimed to quantitatively identify changes in DNA methylation with single site resolution associated with CKD.
Results
Samples
Methylation status was determined for 485 577 unique sites in a total of 407 individuals (cases with CKD, n = 255; controls with no evidence of renal disease n = 152). There were no significant differences for age (36.2 ± 12.0 vs. 36.0 ± 11.8 y) or gender [132 (52%) vs. 76 (50%) males] between case and control groups, respectively, with slightly more males present in the case group. Additional clinical characteristics are provided in Table S1. The 113 individuals with type 1 diabetes had at least 15 y of type 1 diabetes with no evidence of kidney disease (controls) and were compared with 113 individuals with persistent proteinuria and type 1 diabetes (cases) using the same inclusion criteria as our recent meta-analysis of GWAS.Citation10
Quality control
Following stringent quality control and analysis, significantly associated differences in methylation status were observed between 255 individuals with CKD compared with 152 control individuals with no evidence of renal disease. Greater than 99% concordance was observed between duplicate samples (r2 > 0.98 for each of the eight pairs of samples), and experimentally defined genders matched each individual submitted for analysis based on Y-chromosome specific loci.
DNA analysis
Statistically significant association was observed for 399 unique genes in top-ranked markers with P < 10−8. Focusing on top-ranked clusters of associated CpG sites revealed 23 genes, where more than one CpG per gene was identified with Padjusted < 10−8 (; Table S2). Top ranked genes are involved in various diseases, regulation, signaling, and apoptotic pathways (Table S3), and several genes are strong biological candidates for kidney disease including CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2 (Fig. S1). Subgroup analysis of cases and controls without diabetes revealed significant association with the same top-ranked loci as in the original combined analyses. Enrichment based on gene ontology terms revealed 326 processes with P < 0.05 for GO enrichment when considering CpG sites where P < 10−8 for association with CKD; this decreased to 80 when restricted to genes with more than one top-ranked CpG site. Partek pathway analysis identified only three pathways with P < 0.05: mucin type O-Glycan biosynthesis (P = 0.02), DNA replication (P = 0.02), and tryptophan metabolism (P = 0.03) (Tables S4–6). Minimal interactions have been reported between top-ranked genes (Fig. S2).
Table 1. Top-ranked 23 genes showing most significant differences in methylation status between case and control groups
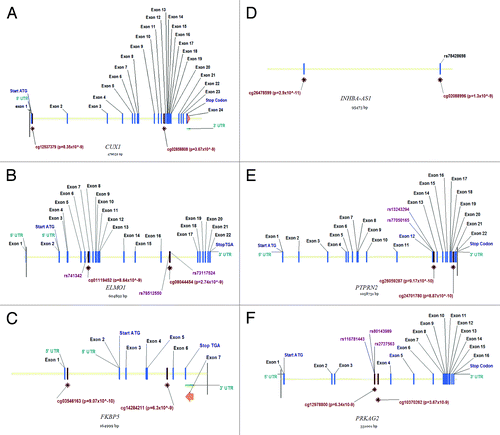
SNPs were identified within the vicinity of CpG sites for key genes (). Fine mapping by genomic and bisulfite treated DNA sequencing confirmed methylation at target sites and also identified two SNPs (rs12673766, PRKAG2, minor allele frequency in our population (9% rather than the 4% reported for CEU population in dbSNP and ELMO1 at 4% minor allele frequency); dbSNP is not presently accepting new SNP submissions so we have included an image of these changes as Figure S3. INHBA-AS1 was not fine-mapped as neither Sanger sequencing (Life Technologies), nor Sequenom Epityper (www.epidesigner.com) primers were successfully designed for bisulfite treated DNA. Examining ENCODE data by means of a regional plot in UCSC’s genome browser for CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2 genes revealed that clusters of CpG sites in PRKAG2 overlapped with a DNaseI hypersensitivity cluster (indicating a gene regulatory region) with ELMO1 and PTPRN2 also having differentially methylated CpG sites in potentially functional sites.
Figure 2. Location of key features for genes that are strong biological candidates for kidney disease including CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2.
RNA analysis
Gene expression, based on total RNA extracted from whole blood, was evaluated by RNA-seq in two cases with CKD stage 5 compared with two controls with no evidence of kidney disease. An average of 71% loading was achieved for chip loading (~70 million reads) with 97% successful enrichment, 24% polyclonal reads for ion sphere particles, and a mean read length of 123 bp. Twenty to 30% polyclonal reads are expected and these were excluded from further analysis. Samples were identified based on their IonXpressRNA barcode and individually analyzed for CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2 genes. None of the key genes demonstrated significant changes in gene expression. However, a non-significant decrease in gene expression was observed for PRKAG2 and a small increase in expression for the ELMO1 gene; of interest, these results do correlate with those expected based on the DNA methylation results. In silico comparisons revealed additional supporting data and some conflicting results (Fig. S4) depending on the source tissue and phenotypes compared for each population; none of this publicly available data examines a CKD population similar to the one investigated in this study.
Several of the loci highlighted in this study were recently identified as differentially methylated genes in kidney tissue [ELMO1 (P = 0.001), PRKAG2 (P = 0.003), CUX1 (P = 0.001), and PTPRN2 (P = 0.005)] comparing 12 individuals with CKD to 14 healthy controls. CUX1 (P = 0.000 000 01) and PTPRN2 (P = 0.000 000 008) genes were also identified in the same paper evaluating methylation profiles between individuals with and without diabetic kidney disease.Citation31
Discussion
Epigenetic modification provides a dynamic link between each individual’s genetic background and relevant environmental exposures. The results described above suggest that adequately powered genome-wide studies of the methylome are both cost-effective and necessary to improve our understanding of CKD. In particular, this novel data suggests that differential methylation in key genes is associated with chronic kidney disease. When not adjusted for age, gender or cell heterogeneity, the majority of differentially methylated sites were hypomethylated. This is not unsurprising as hypomethylation is associated with aging, in particular aging-associated changes in the relative subpopulations of white blood cell types, and age-related disorders.Citation32-Citation34 It is interesting to note that Reinius and colleaguesCitation35 observed that methylation in the promoter-related CpG islands tends to be similar among all white blood cell types so that measurements in whole blood is a reflection of true methylation status across subpopulations at those loci; we observed only 10% of differentially methylated loci in CpG islands (Fig. S5). Following adjustment for age and gender, as well as in silico adjustment for the proportions of white blood cell types using Houseman’s approach,Citation36 many of the hypomethylated regions were not sustained. Hypermethylation of genes has been reported in individuals with ESRDCitation37 and is associated with increased mortality in CKD patients.Citation38 Consistent with these reports we also observed increased DNA methylation in CKD patients (cases) compared with persons without renal disease (controls). However, it must be highlighted that whole-genome methylation is dependent on the method of experimental analysis and that gene-specific methylation depends on the function of the gene under investigation. For example, anticipated methylation differences were observed between case and control groups for pro and anti-inflammatory markers traditionally associated with CKD, but these were not top-ranked in the association tests.
Top-ranked genes have been reported with roles in disease, regulation, signaling, and apoptotic pathways, however formal Partek pathway analysis identified only three pathways with P < 0.05; top-ranked was mucin type O-glycan biosynthesis (P = 0.02) and it is of note that mucin-type O-glycosylation is important for kidney development with abnormalities associated with renal dysfunction, abnormal glomeruli, proximal tubules, and renal podocytes.Citation39-Citation41 Enrichment based on gene ontology also highlighted O-glycosylation with the most significant results being for biological processes protein O-linked glycosylation via serine (P = 0.002, GO ID = 18 242) and protein O-linked glycosylation via threonine (P = 0.002, GO ID = 18 243). Analysis of the interaction profile did not reveal anything of note. Of particular interest are the following genes associated with CKD: CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2. None of the key genes demonstrated significant changes in gene expression based on RNA derived from whole blood, but this is not unexpected as only two case and two control individuals were compared. Nevertheless, it is encouraging that this limited comparison did reveal small changes in gene expression that correlate with our reported DNA methylation status in cases and controls for several genes, including ELMO1 and PRKAG2 in particular. The distribution of methylation in cases and controls for ELMO1 and PRKAG2 is presented in Figure S6.
Comparison with publicly available microarray data sets for gene expression in Nephromine showed conflicting results, although it should be noted that significant differences were observed for PTPRN2, CUX1, and FKBP5 genes in a diabetes cohort comparing 22 microdissected human renal glomerular and tubule samples from healthy patients and from patients with CKD (<60 mL/min/1.73 m2). A recently published paperCitation31 supports association of several of the loci highlighted by our blood-based analysis as ELMO1, PRKAG2, CUX1, and PTPRN2 genes are differentially methylated in kidney tissue comparing individuals with CKD compared with healthy controls.
CUX1 is a member of the homeodomain family of DNA binding proteins that acts as a transcription factor.Citation42 CUX1 is important for kidney development with abnormal expression of CUX1 implicated in kidney disease, particularly polycystic kidney disease.Citation42-Citation44
ELMO1 encodes a member of the engulfment and cell motility protein family and has been associated with diabetic kidney disease.Citation45,Citation46 Decreased methylation was observed in the CKD case group relative to controls and blood-derived gene expression data suggests this may contribute to kidney damage by increasing ELMO1 expression potentially leading to abnormal regulation of the extracellular matrix.Citation47 This decrease in methylation was supported by a higher level of expression for the ELMO1 gene based on RNA-seq results. Increased ELMO1 production may also facilitate more interactions with COX2, thus promoting COX2 activity leading to extracellular matrix accumulation.Citation48
FKBP5 is involved in regulation of the stress hormones and decreased methylation at this FKBP5 locus has been linked to increased stress-dependent gene transcription.Citation49 Decreased methylation was observed in this study, suggesting that FKBP5 may be involved with the development and progression of CKD, as well as the previously reported influence on response to immunosuppressive medication and rejection of kidney transplants.Citation50
INHBA-AS1 encodes a long non-coding antisense RNA potentially involved in kidney damage and transplant rejection.Citation51,Citation52 INHBA acts as a hormone and is involved in growth / differentiation, nephrotoxicity,Citation53 and expression is upregulated in response to immunosuppressive drugs.Citation54
PRKAG2 is located on chromosome 7 and is an important regulator of metabolic functions. Rare genetic variants have been linked to enlarged kidneys and the association of a PRKAG2 SNP (rs7805747, P = 4.2 × 10−12) with CKD was highlighted by Kottgen and colleagues; the CKDGen consortium performed a meta-analysis of genome-wide association data revealing PRKAG2 as one of 13 new loci affecting renal function.Citation55,Citation56
PTPRN2 is an autoantigen associated with type 1 diabetes mellitusCitation57,Citation58 and variation in the PTPRN2 gene has been associated with CKD in Japanese individuals with hypertension and diabetes mellitus.Citation59 SNP rs13243294 in this gene may affect the methylation of cg26059287, but genotypes at this position did not account for the significant difference in methylation in a UK population.
Key strengths of this study include the use of Illumina’s 450K BeadChip, which enables cost-effective, high throughput epigenome-wide association studies examining 485,764 sites distributed across all chromosomes based on selected input from 22 methylation experts across the world.Citation19 Included are unique markers that cover 99% of RefSeq genes with an average of 17 CpG sites per gene region distributed across the promoter, 5′ untranslated region, first exon, gene body, and 3′ untranslated region. This array also includes dedicated content for CpG sites outside CpG islands and microRNA promoter regions.
Similar to our GWAS investigations,Citation10 careful attention was paid to quality control measures for this relatively large-scale epigenetic typing effort. Stringent quality control, consideration of available covariates and strong significance values were employed to minimize false positive associations. Previous methylation studies have shown that < 200 individuals are sufficient to identify associations with common, complex diseasesCitation60-Citation64 and our data supports that proof-of-concept rationale for CKD. This data suggests that larger epigenome-wide association studies of carefully phenotyped individuals may reveal further insights into the biological mechanisms, clinically relevant biomarkers and potential therapeutic options for CKD.
A potential limitation of this study is that blood-derived DNA was used rather than DNA obtained from affected and unaffected kidneys. Laser captured, micro-dissected kidney tissue would allow direct evaluation of methylation chances in relevant tissues; however, this material is not readily available for human population-based studies. Interestingly, increased DNA hypermethylation of RASAL1 appears to influence fibrosis with a model protected from kidney fibrosis following inhibition of DNA methylation.Citation65,Citation66 Kidney biopsies are invasive procedures with attendant surgical risks to the patient, provide a minimal amount of material for analysis, and are not performed routinely unless clinically necessary in the UK. Furthermore, renal tissue within a biopsy sample is heterogeneous consisting of multiple different cell types that make up the microscopic anatomy of the glomerulus, tubule, interstitium, and renal microcirculation. Therefore, the primary aim of our study was to identify biomarkers from a readily accessible source (peripheral blood leucocytes) that may have functional relevance in a routine clinical setting. Differential methylation profiles using blood-derived DNA have previously identified risk factors and biomarkers associated with complex phenotypes.Citation22,Citation28,Citation67-Citation70 A recently published paperCitation31 supports association of several of the loci highlighted by our blood-derived analysis by confirming differentially methylated sites in 2/3 of our top-ranked genes ELMO1, PRKAG2, CUX1, and PTPRN2 genes in kidney tissue (tubular epithelial cells) comparing 12 individuals with CKD to 14 healthy controls. This is a particularly intriguing discovery as it shows the majority of markers identified from peripheral blood were also reflected in kidney specific analysis for CKD.
Recently we identified changes in DNA methylation that were significantly associated with ESRD in our carefully phenotyped, longitudinal renal transplant recipient-donor cohort revealing several biologically relevant pathways under epigenetic control.Citation71 Further large-scale, collaborative endeavors are necessary to characterize genetic and epigenetic risk factors that influence renal function. Meta-analyses are challenging for studies of the methylome due to cross-platform differences and a lack of standardized reporting for methylation studies. Nonetheless, accumulating evidence suggests that DNA methylation differences are associated with complex disease and aggregated results from cross-center studies are revealing promising biomarkers.Citation72,Citation73 The approach described in this manuscript carefully considered tissue type and cell heterogeneity, employed cost-effective 450K analysis for discovery at genome-wide significance, followed by verification and validation using an alternative approach to minimize experimental artifacts, and explored biological relevance alongside expression data for top-ranked markers. Following our identification of associated methylation changes with CKD, replication in additional cohorts is required to confirm these variants and ideally prospective studies would be performed to resolve the methylation influences of cause or effect.
Significant challenges exist in identifying individuals at risk of CKD and optimally managing those diagnosed with CKD in a cost-effective manner.Citation74 Recent studies suggest a complex relationship between genetic variation, DNA methylation and genes expression and it is likely that a model incorporating all of these factors may provide much better biomarkers for CKD. The identification of a genetic-epigenetic profile that can develop understanding of biological mechanisms, lead to earlier diagnosis, and offer improved clinical evaluation would not only provide significant health benefits for affected individuals, but would also have major utility for the efficient use of healthcare resources.
Methods
Samples
All recruited individuals provided written, informed consent and this study was approved by the Office of Research Ethics Northern Ireland. All individuals were from the United Kingdom with White ancestry. Individuals with concurrent disease such as HIV, hepatitis, tuberculosis, lupus or cystic disease were excluded from this study. Cases and controls were carefully selected for age and gender with multivariate regression analysis performed using these covariates for top-ranked loci. DNA was extracted using the salting out method; all samples were treated in a consistent manner, normalized using PicoGreen quantitation, and frozen in multiple aliquots to minimize potential damage from freeze-thaw cycles.
Methylation on 450K
Blood-derived DNA was accurately quantitated using PicoGreen®, normalized, and bisulfite treated using the EZ-96 DNA Methylation-Gold™ Kit (Zymo Research) with case and control samples randomly distributed across arrays. The Infinium Human Methylation 450K BeadChipCitation19,Citation75 (Illumina Inc.) was employed according to manufacturer’s instructions. Raw data were adjusted for dye bias and quantile normalized with data derived from sites using Infinium I or Infinium II assay chemistry considered separately. This high-throughput platform enables quantitative evaluation of methylation levels with single nucleotide resolution, generating a methylation score per individual (a β value ranging from 0 for unmethylated to 1 representing complete methylation) for each CpG site. Standard quality control included evaluation of bisulfite conversion efficiency, staining, hybridization, target removal, extension, dye specificity, and 600 integral negative controls, as well as assessing outliers for gender mismatches, non-White ethnicity and experimental batch effects. All sites with detection P ≥ 0.05 for individual sample’s β value were set to “missing,” as were samples where more than 10% of probes did not generate useful data. Chromosomes X and Y were excluded from association analyses, as were all markers that were ambiguously mapped to the human genome. Normalization of raw methylation data and case-control comparisons for the 450K BeadChip were independently performed using arrayQualityMetrics,Citation76 methylumi (http://tinyurl.com/methylumi), Limma,Citation77 GenomeStudio’s methylation module v1.9 (Illumina) software and customized R scripts; probes were excluded if they failed QC following normalization in any software using standard parameters. Software was used following the developer’s instructions and standard parameters. Formal comparison of normalization methods was not performed, but only those top-ranked sites that were present in independent analyses employing multiple approaches were taken forward for further investigation; we have observed previously that artifacts from the normalization and ranking process (i.e., these are not true methylation changes that can be validated by bisulfite sequencing or Sequenom EpiTyper analysis) are minimized when only those top-ranked loci common to multiple approaches are included. Significance values for differences in methylation levels between groups were adjusted for multiple testing using the Benjamini and Hochberg approach for all probes that passed quality control filters and were included in the association testing for CKD.Citation78 Loci were prioritized where more than one CpG site per gene was identified in top-ranked markers.
In silico analysis
Support for top-ranked genes was sought using published literature and open-access data sources. PubMed (www.ncbi.nlm.nih.gov/pubmed), Scirus (www.scirus.com), Web of Knowledge (www.webofknowledge.com), American Society of Nephrology meeting abstracts (2009–2012), the Renal Association UK abstracts (2009–2012), and Nephromine (www.nephromine.org) were searched using “kidney” or “renal” together with the gene names of primary interest to determine published links. Interactions between top-ranked genes were visualized using STRING 9.05 interaction network.Citation79 NCBI BioSystems DatabaseCitation80 was searched to investigate which pathways were affiliated with top-ranked genes. Illumina’s manifest file was used to filter for known SNPs and dbSNP build 37 was searched for SNPs that may interfere with probe binding. Genotypes for SNP rs13243294 were compared with methylation data for cg26059287 in the PTPRN2 gene from a separate GWAS-EWAS study of 437 UK individuals with type 1 diabetes (McKnight et al., unpublished data/submitted). VectorNTI software (Life Technologies) was employed to visualize the location of features for genes () with probe sequences, target CpG sites and local SNPs highlighted (Fig. S2). Gene set enrichment and pathway analysis was conducted using Partek Genomics Suite 6.6.
Nephromine™ (Compendia Bioscience™, part of Life Technologies™ www.nephromine.org) was used for analysis and visualization of in silico gene expression data sets.
Fine mapping
Primers were designed to fine map the regions immediately surrounding top-ranked CpG sites in CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2 genes (Fig. S7; Table S7). Forty-six samples (23 cases and 23 controls) were bidirectionally analyzed for SNPs using Sanger sequencing on a 3730 genetic analyzer (Life Technologies) as this provides 95% power to detect all polymorphisms with greater than 5% minor allele frequency.Citation81 Methylation was confirmed using bisulfite treated DNA compared with genomic DNA for the same individuals.
RNA-Seq
To evaluate the relationship between gene expression and methylation status for CUX1, ELMO1, FKBP5, INHBA-AS1, PTPRN2, and PRKAG2 genes, blood was collected within 5 min in both EDTA and PAX gene tubes and processed for DNA and RNA extraction for four individuals; two cases with CKD and two controls with no evidence of kidney disease. RNA was extracted from 2.5 ml of whole blood using the PAXgene RNA System following manufacturer’s instructions. RNA was selectively depleted for rRNA using the RiboMinus™ Eukaryote System v2 (Life Technologies) and libraries were prepared according to the Ion Total RNA-Seq Kit v2 for Whole Transcriptome Libraries standard protocol, with samples barcoded using RNA-Seq Library Barcodes 1–4 (Life Technologies). Reference spike-in controls from the External RNA Controls Consortium (ERCC) were included for quality control purposes. Prepared libraries were diluted to 12 pM using TE 0.1 and enriched using a One-Touch Two machine and automated enrichment system with the Ion PI™ Template OT2 200 Kit version two. Enriched products were prepared for sequencing using the PI™ Sequencing 200 Kit version two and run on a Proton using version two of the P1 chip (Life Technologies). Data was analyzed using Torrent Suite v3.6, Partek Flow and key genes examined using the template workflow in Partek Genomics Suite 6.6.
Additional data are available from the authors or at www.qub.ac.uk/neph-res/CORGI.
| Abbreviations: | ||
| CKD | = | chronic kidney disease |
| eGFR | = | estimated glomerular filtration rate |
| ESRD | = | end stage renal disease |
Additional material
Download Zip (1.1 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Funding was provided by the Northern Ireland Research and Development Office, and the Northern Ireland Kidney Research Fund. Smyth LJ is supported by a PhD studentship from the Northern Ireland Department for Employment and Learning.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/27161
References
- Levey AS, Coresh J. Chronic kidney disease. Lancet 2012; 379:165 - 80; http://dx.doi.org/10.1016/S0140-6736(11)60178-5; PMID: 21840587
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380:2095 - 128; http://dx.doi.org/10.1016/S0140-6736(12)61728-0; PMID: 23245604
- McCullough K, Sharma P, Ali T, Khan I, Smith WC, MacLeod A, Black C. Measuring the population burden of chronic kidney disease: a systematic literature review of the estimated prevalence of impaired kidney function. Nephrol Dial Transplant 2012; 27:1812 - 21; http://dx.doi.org/10.1093/ndt/gfr547; PMID: 21965592
- Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003; 41:1 - 12; http://dx.doi.org/10.1053/ajkd.2003.50007; PMID: 12500213
- Castledine C, Casula A, Fogarty D. Chapter 2 UK RRT prevalence in 2010: national and centre-specific analyses. Nephron Clin Pract 2012; 120:Suppl 1 c29 - 54; http://dx.doi.org/10.1159/000342844; PMID: 22964573
- Kerr M, Bray B, Medcalf J, O’Donoghue DJ, Matthews B. Estimating the financial cost of chronic kidney disease to the NHS in England. Nephrol Dial Transplant 2012; 27:Suppl 3 iii73 - 80; http://dx.doi.org/10.1093/ndt/gfs269; PMID: 22815543
- Levey AS, Coresh J. Chronic kidney disease. Lancet 2012; 379:165 - 80; http://dx.doi.org/10.1016/S0140-6736(11)60178-5; PMID: 21840587
- Chasman DI, Fuchsberger C, Pattaro C, Teumer A, Böger CA, Endlich K, Olden M, Chen MH, Tin A, Taliun D, et al, CARDIoGRAM Consortium, ICBP Consortium, CARe Consortium, WTCCC2. Integration of genome-wide association studies with biological knowledge identifies six novel genes related to kidney function. Hum Mol Genet 2012; 21:5329 - 43; http://dx.doi.org/10.1093/hmg/dds369; PMID: 22962313
- Böger CA, Gorski M, Li M, Hoffmann MM, Huang C, Yang Q, Teumer A, Krane V, O’Seaghdha CM, Kutalik Z, et al, CKDGen Consortium. Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD. PLoS Genet 2011; 7:e1002292; http://dx.doi.org/10.1371/journal.pgen.1002292; PMID: 21980298
- Sandholm N, Salem RM, McKnight AJ, Brennan EP, Forsblom C, Isakova T, McKay GJ, Williams WW, Sadlier DM, Mäkinen VP, et al, DCCT/EDIC Research Group. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet 2012; 8:e1002921; http://dx.doi.org/10.1371/journal.pgen.1002921; PMID: 23028342
- McKnight AJ, Currie D, Maxwell AP. Unravelling the genetic basis of renal diseases; from single gene to multifactorial disorders. J Pathol 2010; 220:198 - 216; PMID: 19882676
- McKnight AJ, Maxwell AP. Bioinformatic resources for diabetic nephropathy. J Diabetes and Bioinformatics 2013; In press
- Keller BJ, Martini S, Sedor JR, Kretzler M. A systems view of genetics in chronic kidney disease. Kidney Int 2012; 81:14 - 21; http://dx.doi.org/10.1038/ki.2011.359; PMID: 22012128
- Dwivedi RS, Herman JG, McCaffrey TA, Raj DS. Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int 2011; 79:23 - 32; http://dx.doi.org/10.1038/ki.2010.335; PMID: 20881938
- Ekström TJ, Stenvinkel P. The epigenetic conductor: a genomic orchestrator in chronic kidney disease complications?. J Nephrol 2009; 22:442 - 9; PMID: 19662598
- McCaughan JA, McKnight AJ, Courtney AE, Maxwell AP. Epigenetics: time to translate into transplantation. Transplantation 2012; 94:1 - 7; http://dx.doi.org/10.1097/TP.0b013e31824db9bd; PMID: 22437848
- Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA 2008; 299:1345 - 50; http://dx.doi.org/10.1001/jama.299.11.1345; PMID: 18349095
- Flintoft L. Disease epigenomics: a smoking gun. Nat Rev Genet 2011; 12:300; http://dx.doi.org/10.1038/nrg2991; PMID: 21483458
- Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011; 12:529 - 41; http://dx.doi.org/10.1038/nrg3000; PMID: 21747404
- Dwivedi RS, Herman JG, McCaffrey TA, Raj DS. Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int 2011; 79:23 - 32; http://dx.doi.org/10.1038/ki.2010.335; PMID: 20881938
- Reddy MA, Natarajan R. Epigenetics in diabetic kidney disease. J Am Soc Nephrol 2011; 22:2182 - 5; http://dx.doi.org/10.1681/ASN.2011060629; PMID: 22021712
- Chowdhury S, Erickson SW, MacLeod SL, Cleves MA, Hu P, Karim MA, Hobbs CA. Maternal genome-wide DNA methylation patterns and congenital heart defects. PLoS One 2011; 6:e16506; http://dx.doi.org/10.1371/journal.pone.0016506; PMID: 21297937
- Luttropp K, Lindholm B, Carrero JJ, Glorieux G, Schepers E, Vanholder R, Schalling M, Stenvinkel P, Nordfors L. Genetics/Genomics in chronic kidney disease--towards personalized medicine?. Semin Dial 2009; 22:417 - 22; http://dx.doi.org/10.1111/j.1525-139X.2009.00592.x; PMID: 19708993
- Joss-Moore LA, Lane RH. Epigenetics and the developmental origins of disease: the key to unlocking the door of personalized medicine. Epigenomics 2012; 4:471 - 3; http://dx.doi.org/10.2217/epi.12.53; PMID: 23130826
- Liakopoulos V, Georgianos PI, Eleftheriadis T, Sarafidis PA. Epigenetic mechanisms and kidney diseases. Curr Med Chem 2011; 18:1733 - 9; http://dx.doi.org/10.2174/092986711795496827; PMID: 21466478
- Tsai PC, Spector TD, Bell JT. Using epigenome-wide association scans of DNA methylation in age-related complex human traits. Epigenomics 2012; 4:511 - 26; http://dx.doi.org/10.2217/epi.12.45; PMID: 23130833
- Syreeni A, El-Osta A, Forsblom C, Sandholm N, Parkkonen M, Tarnow L, Parving HH, McKnight AJ, Maxwell AP, Cooper ME, et al, FinnDiane Study Group. Genetic examination of SETD7 and SUV39H1/H2 methyltransferases and the risk of diabetes complications in patients with type 1 diabetes. Diabetes 2011; 60:3073 - 80; http://dx.doi.org/10.2337/db11-0073; PMID: 21896933
- Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics 2010; 3:33; http://dx.doi.org/10.1186/1755-8794-3-33; PMID: 20687937
- Young GH, Wu VC. KLOTHO methylation is linked to uremic toxins and chronic kidney disease. Kidney Int 2012; 81:611 - 2; http://dx.doi.org/10.1038/ki.2011.461; PMID: 22419041
- Sapienza C, Lee J, Powell J, Erinle O, Yafai F, Reichert J, Siraj ES, Madaio M. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics 2011; 6:20 - 8; http://dx.doi.org/10.4161/epi.6.1.13362; PMID: 21150313
- Ko YA, Mohtat D, Suzuki M, Park ASD, Izquierdo MC, Han SY, Kang HM, Si H, Hostetter T, Pullman JM, et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol 2013; 14:R108; http://dx.doi.org/10.1186/gb-2013-14-10-r108; PMID: 24098934
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 2010; 20:434 - 9; http://dx.doi.org/10.1101/gr.103101.109; PMID: 20219945
- Ong ML, Holbrook JD. Novel region discovery method for Infinium 450K DNA methylation data reveals changes associated with aging in muscle and neuronal pathways. Aging Cell 2013; Forthcoming http://dx.doi.org/10.1111/acel.12159; PMID: 24112369
- Pobribny IP, Vanyushin BF. Age related genomic hypomethylation in epigenetics of aging. Epigenetics of Aging (ed Tollefsbol TO); 2010. ISBN 978—1-4419-0638-0.
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, Söderhäll C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012; 7:e41361; http://dx.doi.org/10.1371/journal.pone.0041361; PMID: 22848472
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13:86; http://dx.doi.org/10.1186/1471-2105-13-86; PMID: 22568884
- Geisel J, Schorr H, Heine GH, Bodis M, Hübner U, Knapp JP, Herrmann W. Decreased p66Shc promoter methylation in patients with end-stage renal disease. Clin Chem Lab Med 2007; 45:1764 - 70; http://dx.doi.org/10.1515/CCLM.2007.357; PMID: 18067454
- Stenvinkel P, Karimi M, Johansson S, Axelsson J, Suliman M, Lindholm B, Heimbürger O, Barany P, Alvestrand A, Nordfors L, et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease?. J Intern Med 2007; 261:488 - 99; http://dx.doi.org/10.1111/j.1365-2796.2007.01777.x; PMID: 17444888
- Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, Shinzato T, Kobayashi Y, Maeda K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int 2001; 59:1077 - 85; http://dx.doi.org/10.1046/j.1523-1755.2001.0590031077.x; PMID: 11231363
- Tian E, Ten Hagen KG. Recent insights into the biological roles of mucin-type O-glycosylation. Glycoconj J 2009; 26:325 - 34; http://dx.doi.org/10.1007/s10719-008-9162-4; PMID: 18695988
- Kakani S, Yardeni T, Poling J, Ciccone C, Niethamer T, Klootwijk ED, Manoli I, Darvish D, Hoogstraten-Miller S, Zerfas P, et al. The Gne M712T mouse as a model for human glomerulopathy. Am J Pathol 2012; 180:1431 - 40; http://dx.doi.org/10.1016/j.ajpath.2011.12.023; PMID: 22322304
- Alcalay NI, Vanden Heuvel GB. Regulation of cell proliferation and differentiation in the kidney. Front Biosci (Landmark Ed) 2009; 14:4978 - 91; http://dx.doi.org/10.2741/3582; PMID: 19482600
- Hulea L, Nepveu A. CUX1 transcription factors: from biochemical activities and cell-based assays to mouse models and human diseases. Gene 2012; 497:18 - 26; http://dx.doi.org/10.1016/j.gene.2012.01.039; PMID: 22306263
- Fragiadaki M, Ikeda T, Witherden A, Mason RM, Abraham D, Bou-Gharios G. High doses of TGF-β potently suppress type I collagen via the transcription factor CUX1. Mol Biol Cell 2011; 22:1836 - 44; http://dx.doi.org/10.1091/mbc.E10-08-0669; PMID: 21471005
- Shimazaki A, Kawamura Y, Kanazawa A, Sekine A, Saito S, Tsunoda T, Koya D, Babazono T, Tanaka Y, Matsuda M, et al. Genetic variations in the gene encoding ELMO1 are associated with susceptibility to diabetic nephropathy. Diabetes 2005; 54:1171 - 8; http://dx.doi.org/10.2337/diabetes.54.4.1171; PMID: 15793258
- Hanson RL, Millis MP, Young NJ, Kobes S, Nelson RG, Knowler WC, DiStefano JK. ELMO1 variants and susceptibility to diabetic nephropathy in American Indians. Mol Genet Metab 2010; 101:383 - 90; http://dx.doi.org/10.1016/j.ymgme.2010.08.014; PMID: 20826100
- Shimazaki A, Tanaka Y, Shinosaki T, Ikeda M, Watada H, Hirose T, Kawamori R, Maeda S. ELMO1 increases expression of extracellular matrix proteins and inhibits cell adhesion to ECMs. Kidney Int 2006; 70:1769 - 76; http://dx.doi.org/10.1038/sj.ki.5001939; PMID: 17021600
- Yang C, Sorokin A. Upregulation of fibronectin expression by COX-2 is mediated by interaction with ELMO1. Cell Signal 2011; 23:99 - 104; http://dx.doi.org/10.1016/j.cellsig.2010.08.008; PMID: 20732417
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS, Bradley B, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 2013; 16:33 - 41; http://dx.doi.org/10.1038/nn.3275; PMID: 23201972
- Grigoryev DN, Liu M, Cheadle C, Barnes KC, Rabb H. Genomic profiling of kidney ischemia-reperfusion reveals expression of specific alloimmunity-associated genes: Linking “immune” and “nonimmune” injury events. Transplant Proc 2006; 38:3333 - 6; http://dx.doi.org/10.1016/j.transproceed.2006.10.129; PMID: 17175265
- Happé H, van der Wal AM, Leonhard WN, Kunnen SJ, Breuning MH, de Heer E, Peters DJ. Altered Hippo signalling in polycystic kidney disease. J Pathol 2011; 224:133 - 42; http://dx.doi.org/10.1002/path.2856; PMID: 21381034
- Famulski KS, Sis B, Billesberger L, Halloran PF. Interferon-gamma and donor MHC class I control alternative macrophage activation and activin expression in rejecting kidney allografts: a shift in the Th1-Th2 paradigm. Am J Transplant 2008; 8:547 - 56; http://dx.doi.org/10.1111/j.1600-6143.2007.02118.x; PMID: 18294151
- Garrett SH, Clarke K, Sens DA, Deng Y, Somji S, Zhang KK. Short and long term gene expression variation and networking in human proximal tubule cells when exposed to cadmium. BMC Med Genomics 2013; 6:Suppl 1 S2; http://dx.doi.org/10.1186/1755-8794-6-S1-S2; PMID: 23369406
- Wen J, Ji ZG, Niu JR. [Regulatory effects of cyclosporin a and tacrolimus on th immunological gene expressions in renal transplant recipients]. [Abstract only] Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2012; 34:563 - 6; PMID: 23286399
- Böger CA, Gorski M, Li M, Hoffmann MM, Huang C, Yang Q, Teumer A, Krane V, O’Seaghdha CM, Kutalik Z, et al, CKDGen Consortium. Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD. PLoS Genet 2011; 7:e1002292; http://dx.doi.org/10.1371/journal.pgen.1002292; PMID: 21980298
- Köttgen A, Pattaro C, Böger CA, Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith AV, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet 2010; 42:376 - 84; http://dx.doi.org/10.1038/ng.568; PMID: 20383146
- Wasmeier C, Hutton JC. Molecular cloning of phogrin, a protein-tyrosine phosphatase homologue localized to insulin secretory granule membranes. J Biol Chem 1996; 271:18161 - 70; http://dx.doi.org/10.1074/jbc.271.30.18161; PMID: 8663434
- Kawasaki E, Hutton JC, Eisenbarth GS. Molecular cloning and characterization of the human transmembrane protein tyrosine phosphatase homologue, phogrin, an autoantigen of type 1 diabetes. Biochem Biophys Res Commun 1996; 227:440 - 7; http://dx.doi.org/10.1006/bbrc.1996.1526; PMID: 8878534
- Yoshida T, Kato K, Yokoi K, Oguri M, Watanabe S, Metoki N, Yoshida H, Satoh K, Aoyagi Y, Nozawa Y, et al. Association of genetic variants with chronic kidney disease in Japanese individuals with or without hypertension or diabetes mellitus. Exp Ther Med 2010; 1:137 - 45; PMID: 23136606
- Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, Daunay A, Busato F, Mein CA, Manfras B, et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 2011; 7:e1002300; http://dx.doi.org/10.1371/journal.pgen.1002300; PMID: 21980303
- Philibert RA, Plume JM, Gibbons FX, Brody GH, Beach SR. The impact of recent alcohol use on genome wide DNA methylation signatures. Front Genet 2012; 3:54; http://dx.doi.org/10.3389/fgene.2012.00054; PMID: 22514556
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88:450 - 7; http://dx.doi.org/10.1016/j.ajhg.2011.03.003; PMID: 21457905
- Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis 2013; 73:110 - 7; http://dx.doi.org/10.1136/annrheumdis-2012-201526; PMID: 22736089
- Harris RA, Nagy-Szakal D, Pedersen N, Opekun A, Bronsky J, Munkholm P, Jespersgaard C, Andersen P, Melegh B, Ferry G, et al. Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis 2012; 18:2334 - 41; http://dx.doi.org/10.1002/ibd.22956; PMID: 22467598
- Bechtel W, McGoohan S, Zeisberg EM, Müller GA, Kalbacher H, Salant DJ, Müller CA, Kalluri R, Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med 2010; 16:544 - 50; http://dx.doi.org/10.1038/nm.2135; PMID: 20418885
- Woroniecki R, Gaikwad AB, Susztak K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr Nephrol 2011; 26:705 - 11; http://dx.doi.org/10.1007/s00467-010-1714-8; PMID: 21174217
- Flintoft L. Disease epigenomics: a smoking gun. Nat Rev Genet 2011; 12:300; http://dx.doi.org/10.1038/nrg2991; PMID: 21483458
- Harris RA, Nagy-Szakal D, Pedersen N, Opekun A, Bronsky J, Munkholm P, Jespersgaard C, Andersen P, Melegh B, Ferry G, et al. Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis 2012; 18:2334 - 41; http://dx.doi.org/10.1002/ibd.22956; PMID: 22467598
- Zhu Y, Stevens RG, Hoffman AE, Tjonneland A, Vogel UB, Zheng T, Hansen J. Epigenetic impact of long-term shiftwork: pilot evidence from circadian genes and whole-genome methylation analysis. Chronobiol Int 2011; 28:852 - 61; http://dx.doi.org/10.3109/07420528.2011.618896; PMID: 22080730
- Wang L, Aakre JA, Jiang R, Marks RS, Wu Y, Chen J, Thibodeau SN, Pankratz VS, Yang P. Methylation markers for small cell lung cancer in peripheral blood leukocyte DNA. J Thorac Oncol 2010; 5:778 - 85; http://dx.doi.org/10.1097/JTO.0b013e3181d6e0b3; PMID: 20421821
- McCaughan JA, Courtney AE, Maxwell AP, McKnight AJ. Genome wide methylation analysis of 485,577 features in a renal transplant population. J Am Soc Nephrol 2011; 22:458A
- Sapari NS, Loh M, Vaithilingam A, Soong R. Clinical potential of DNA methylation in gastric cancer: a meta-analysis. PLoS One 2012; 7:e36275; http://dx.doi.org/10.1371/journal.pone.0036275; PMID: 22558417
- Woo HD, Kim J. Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One 2012; 7:e34615; http://dx.doi.org/10.1371/journal.pone.0034615; PMID: 22509334
- Black C, Sharma P, Scotland G, McCullough K, McGurn D, Robertson L, Fluck N, MacLeod A, McNamee P, Prescott G, et al. Early referral strategies for management of people with markers of renal disease: a systematic review of the evidence of clinical effectiveness, cost-effectiveness and economic analysis. Health Technol Assess 2010; 14:1 - 184; PMID: 20441712
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- Kauffmann A, Huber W. Microarray data quality control improves the detection of differentially expressed genes. Genomics 2010; 95:138 - 42; http://dx.doi.org/10.1016/j.ygeno.2010.01.003; PMID: 20079422
- Smyth G. Limma: linear models for microarray data in Bioinformatics and Computational Biology Solutions using R and Bioconductor 397-420. Springer, New York (2005).
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc, B 1995; 57:289 - 300
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013; 41:D808 - 15; http://dx.doi.org/10.1093/nar/gks1094; PMID: 23203871
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2013; 41:D8 - 20; http://dx.doi.org/10.1093/nar/gks1189; PMID: 23193264
- McKnight AJ, Woodman AM, Parkkonen M, Patterson CC, Savage DA, Forsblom C, Pettigrew KA, Sadlier D, Groop PH, Maxwell AP, Warren 3/UK GoKinD Study Group. Investigation of DNA polymorphisms in SMAD genes for genetic predisposition to diabetic nephropathy in patients with type 1 diabetes mellitus. Diabetologia 2009; 52:844 - 9; http://dx.doi.org/10.1007/s00125-009-1281-3; PMID: 19247629