Abstract
Antigen/antibody complexes can efficiently target antigen presenting cells to allow stimulation of the cellular immune response. Due to the difficulty of manufacture and their inherent instability complexes have proved inefficient cancer vaccines. However, anti-idiotypic antibodies mimicking antigens have been shown to stimulate both antibody and T cell responses. The latter are due to T cell mimotopes expressed within the complementarity-determining regions (CDRs) of antibodies that are efficiently presented to dendritic cells in vivo. Based on this observation we have designed a DNA vaccine platform called ImmunoBodyTM, where cytotoxic T lymphocyte (CTL) and helper T cell epitopes replace CDR regions within the framework of a human IgG1 antibody. The ImmunoBodyTM expression system has a number of design features which allow for rapid production of a wide range of vaccines. The CDR regions of the heavy and light chain have been engineered to contain unique restriction endonuclease sites, which can be easily opened, and oligonucleotides encoding the T cell epitopes inserted. The variable and constant regions of the ImmunoBodyTM are also flanked by restriction sites, which permit easy exchange of other IgG subtypes. Here we show a range of T cell epitopes can be inserted into the ImmunoBodyTM vector and upon immunization these T cell epitopes are efficiently processed and presented to stimulate high frequency helper and CTL responses capable of anti-tumor activity.
Introduction
The aim of a cancer vaccine is to stimulate anti-tumor cellular immunity by inducing T cell responses. In order for this to be achieved, antigen presenting cells such as dendritic cells (DCs) process the vaccine and present epitopes on either MHC class I or MHC class II molecules, which react with the T cell receptor (TCR) of CD8 or CD4 T cells, respectively. T cells recognizing these complexes can then be activated as amplifiers of the response (CD4 helper T cells) or as direct effectors (CD8 cytotoxic T lymphocytes) to attack the tumor cells. Stimulation of T cell responses also has implications for traditional monoclonal antibody (mAb) therapy as presentation of antibody/antigen immune complexes including tumor cells opsonized with mAb, could lead to cellular immune responses.Citation1 Indeed, it is such effects that are sometimes evoked to explain the long term responses observed in lymphoma patients after therapy with the anti-CD20 antibody Rituximab (Rituxan).Citation2
Immune complexes destined for immunization are notoriously difficult to manufacture and have low stability in vivo. However, anti-idiotypic antibodies can act like antigen/antibody complexes where the antigenic epitope is encoded within the variable region of the antibody whilst retaining the in vivo stability of monomeric antibody. Anti-idiotypic antibodies mimicking antigens have been shown to stimulate both antibody and T cell responses.Citation3 The latter are due to T cell mimotopes expressed within their complementarity-determining regions (CDRs) which are processed and presented on MHC antigens and stimulate a T cell response which can recognize the wild type epitope processed from the mimicked antigen.Citation4 We have previously shown that a human monoclonal IgG1 anti-idiotypic antibody, 105AD7, isolated from a colorectal cancer patient who survived seven years with extensive liver metastases, can stimulate helper and cytotoxic T cell responses in over 300 cancer patients with no associated toxicity.Citation5–Citation22
Isolating anti-idiotypic antibodies expressing T cell mimotopes is difficult. However, it is possible to genetically modify antibodies to express B cell and T cell epitopes within their CDR regions.Citation23,Citation24 The three CDR loops of the heavy and light chain variable regions interconnect β strands of conserved framework regions. All CDRs show a high level of sequence diversity and form a binding surface for antigen interaction.Citation25,Citation26 However, within their sequence there are a number of key residues present that make contact with portions of the framework or have side chains buried within the hydrophobic core.Citation27–Citation31 It is anticipated that removal of the CDR and its replacement with epitopes would have a significant impact on folding and stability of the antibody. With the exception of H3, all CDRs are canonical in structures that are generally conserved in length and are involved in variable heavy and light chain scaffolding.Citation27,Citation32–Citation34 However, CDR3 is much more diverse both in sequence variability and in length.Citation32,Citation33 Replacement of CDR3 tends to have less impact on folding and secretion.Citation35 We therefore made a series of DNA vaccines by substituting a number of CTL and helper epitopes within different CDRs and measured their ability to secrete antibody and stimulate T cell responses. This report shows that a DNA vaccine encoding T cell epitopes within an antibody framework is an efficient method for stimulating T cell responses and generating a significant delay in tumor growth.
Results
To allow rapid development of antibodies expressing known T cell epitopes a vector expressing a human IgG1 antibody with its CDRs replaced by restriction sites was generated ().
Replacement of CDRs with unique restriction recognition sequences.
With exception of the heavy CDR2 region that retains six amino acids, the CDRs of the deimmunized VH and VL chains were completely removed and exchanged for unique restriction enzyme sites. Selection of these sites was crucial as each site had to be unique for each CDR and not be present in the rest of the vector. lists chosen enzyme sites for all CDRs. These unique restriction sites were used to open up the DNA such that an oligonucleotide encoding an antigenic epitope could be inserted. Most framework sequence that was lost on generation of the restriction site was subsequently replaced by including these residues with the inserted epitope. Either one or more CDR regions were removed and replaced allowing generation of vectors that would allow single or multiple epitopes (from the same or different antigens) to be inserted.
Insertion of antigenic epitopes into CDR sites of single chain vectors.
Epitopes can easily be inserted into any of the sites within the single chain vectors. A number of CD8 CTL and CD4 helper epitopes were inserted into the ImmunoBody™ vectors (). Complementary oligonucleotides were designed to encode nucleotide sequence that on translation express the epitope (). Epitope insertions were confirmed by sequencing within the single vectors using the universal primer CMV forward. Once all epitopes have been incorporated into the VH and VL sites within the single vectors, they were transferred into the double expression vector pDCOrig () using HindIII/Afe1 and BamHI/BsiWI in frame with their respective human constant regions. pDCOrig contains both the heavy and light chain gene coding sequences combined within the same construct. Careful design of this vector has retained the unique restriction enzyme sites at the junctions of the variable and constant regions and provided a quick and easy method to create different combinations of the variable regions. These constructs were initially screened for protein secretion.
Protein secretion from CDR substituted antibodies.
A range of helper and cytotoxic T cell epitopes were cloned into the different CDR regions of the ImmunoBody™ vector. To determine if the ImmunoBody™ constructs secreted intact, heavy or light chains CHO-S cells were transfected and tested for Ig secretion by ELISA (). Supernatants and corresponding purified protein were analyzed by western blotting ( and B). Purified intact antibody was apparent in purified and supernatant from all three constructs ( and B; lanes 1, 2, 3, 4, 5 and 6 respectively). However larger amounts of free heavy chain were detected when TRP2 was grafted into CDRH2 alongside gp100 210M in CDRH1 and the CD4 HepB helper epitope in CDRL1 ( lane 3; B lanes 3 and 4). Secretion of intact antibody was also detected by ELISA () from the wild type ImmunoBody™ containing native CDRs as well as those substituted with CTL or helper epitopes in CDRH3 or CTL epitopes within CDRL3. In contrast substitution of helper or CTL epitopes in CDRL1, CDRH1 or CDRH2 demonstrated very little intact antibody or light chain but secretion of large amounts of the heavy chain.
To investigate whether multiple epitopes could be inserted into the CDRH3 site and still retain folding, HepB helper or TRP2 CTL epitope repeats linked by triple alanines of varying lengths and hydropathicity were incorporated into this CDR. Hydropathicity plots show that the hydropathic profile of the wild type sequence in CDRH3 was maintained with replacement of the HepB epitope repeat in contrast to that of the TRP2 epitope repeat (). In line with the hydropathicity prediction, secretion of intact antibody was maintained on insertion of the HepB epitope repeat of 29 amino acids, but not with insertion of the TRP2 epitope repeat of 21 amino acids (). All constructs whether they produced large amounts of intact antibody or not were then assessed as DNA vaccines for their ability to stimulate CTL and helper responses.
Large amounts of intact antibody is not required to stimulate T cell responses.
As DNA can stimulate immune response by direct presentation (transfection of antigen presenting cells) or by cross presentation (secretion of protein which is then taken up by antigen presenting cells), it was of interest to assess if secretion of intact antibody was required to stimulate T cell responses. A construct containing the TRP-2 H-2Kb epitope in the CDRH3, which was secreted as intact antibody, was compared to a construct containing the same epitope in CDRH2, and which mainly secreted heavy chain. Both DNA vaccines were also compared to an intact protein version of the ImmunoBody™ vaccine containing the epitope in CDRH3. C57Bl/6 mice were immunized three times at weekly intervals and splenocytes analyzed for the presence of epitope specific immune responses ex vivo by IFNγ elispot assay. The DNA construct that only secreted low amounts of intact antibody and high levels of free heavy chain had a significantly higher frequency T cell response compared to the DNA construct encoding intact antibody (p = 0.0011; ). However, both the DNA vaccines elicit higher frequency responses compared to mice immunized with the ImmunoBody™ protein vaccine (p < 0.0001 and p = 0.0028). This may be due to the DNA vaccine stimulating immune responses by direct presentation, which does not rely on protein secretion. The leader sequence was therefore removed from the ImmunoBody™ expressing the TRP-2 epitope within its CDRH2. Unexpectedly this resulted in a significant reduction in the CTL response (p = 0.0085), suggesting that direct presentation was not solely responsible for stimulating CTL responses and secretion of at least heavy chain is an advantage (). The DNA could also be superior to the protein vaccine as it contains immunostimulatory CpG sites, which would also be true for a DNA vaccine encoding antigen.
ImmunoBody™ DNA immunization elicits higher frequency responses than immunization with whole antigen DNA.
Immunization with antigen may provide more efficient processing and presentation compared to epitope encoded within an antibody molecule. The ImmunoBody™ construct containing the H-2Kb restricted TRP2 epitope in CDRH2 was therefore compared to DNA expressing whole murine TRP2 antigen. Immunization of C57Bl6 mice revealed significantly higher frequency responses elicited from the ImmunoBody™ DNA compared to the whole antigen DNA in pcDNA3 vector (p = 0.0034) (). To prove that the lack of response to whole antigen TRP2 was not due to the vector backbone, the complete murine TRP2 antigen sequence was engineered into the same expression vector as the ImmunoBody™ construct. The change in vector backbone still failed to induce a response from whole TRP2 antigen comparable to that observed with ImmunoBody™ DNA (p = 0.0055). These results suggest that an ImmunoBody™ expressing TRP-2 epitope is more efficient at stimulating CTL responses than a DNA vaccine encoding antigen, possibly due to less competing epitopes within the inert antibody carrier. To ensure that the optimal response was being stimulated by the ImmunoBody™ vaccine single and multiple immunizations were compared.
ImmunoBody™ DNA immunization requires a prime boost regime for generation of optimal responses.
The majority of immunization regimes to date involve the use of prime and boost schedules which are believed to induce better immune responses. To address this in the context of the ImmunoBody™ DNA vaccine, C57Bl/6 mice were given a single immunization and responses were analyzed at 7, 14 or 20 days post immunization. No epitope specific responses were observed (data not shown); therefore it was deduced that a prime and boost would be necessary. C57Bl6 mice were subsequently immunized at days 0 and 7 or 0, 7 and 14 with ImmunoBody™ DNA, and epitope specific responses were analyzed at day 20 post primary immunization by IFNγ elispot assay. demonstrates that responses were detectable after two immunizations although these were not significant over control. Frequency was significantly increased after three immunizations compared to control (p = 0.0043), and this was significantly better than a single immunization (p = 0.006). As three immunizations led to generation of the strongest immune response, the persistence of this response was assessed using this regime. C57Bl/6 mice were immunized three times with ImmunoBody™ DNA and immune responses analyzed at day 20 and day 48 post primary immunization. A parallel group was given a booster at day 42 post primary immunization. High frequency responses observed at day 20 post primary immunization (p = 0.0043) were dramatically reduced in frequency at day 48, but were still significant compared to control (p = 0.0024). However, administration of a booster immunization at day 42 induced rapid expansion and recovery of the response (). Although strong CTL responses were stimulated by ImmunoBody™ vaccination, it was unclear if CD4 help would enhance these responses.
Does human Fc provide xenogenic help?
Although ImmunoBody™ was very efficient at stimulating CTL responses, it was unclear if these responses required linked CD4 help. Mice were therefore immunized with a DNA vector encoding only heavy chain containing either H-2Kb restricted TRP2 epitope or HLA-A2 restricted gp100 210M epitope. High frequency CTL responses were still generated from both constructs compared to control (p = 0.0031 and p = 0.0004 respectively; ). This implied that either CD4 help is not required or that the human Fc region which is xenogenic in mice is providing linked foreign help.
The ImmunoBody™ vector design allows for the IgG constant region to be removed by a simple restriction digest and replacement with different IgG constant regions. The human IgG1 and kappa constant regions of the ImmunoBody™ encoding the H-2Kb restricted TRP-2 epitope in its CDRH2 and the I-Ab restricted HepB epitope in its CDR1L1 was replaced with the equivalent murine IgG2a regions. This construct was assessed for secretion of heavy and light chains, and demonstrated identical secretion patterns to the human IgG1 equivalent (DCIB18; ). The murine IgG2a construct was screened for generation of immune responses. Although this construct still gave high frequency CTL and helper responses, these were not as strong as responses from the equivalent human construct (p = 0.0003), suggesting that the xenogenic Fc was providing help (). An HLA-DR4 restricted gp100 epitope was then incorporated into the mouse IgG2a construct to provide linked help for CTL generation. This construct elicited high frequency CD8 and CD4 epitope specific responses () in HLA-DR4 transgenic mice that were comparable to those elicited by the human IgG1 construct. These results demonstrate that CTL responses can be stimulated in the absence of linked CD4 help, but that its presence is required for optimal responses. However, strong helper responses can also be generated from epitopes incorporated within the light chain. As antigen specific help is required to set up chemokine gradients within tumors to allow extravasation of CTL responses, it may be preferable to engineer helper epitopes within both the heavy and light chains. Multiple helper epitopes where therefore cloned into different CDR sites within ImmunoBody™ and screened for their ability to stimulate helper responses.
Helper responses can be generated from different epitopes cloned within the CDRL1 or CDRL3 region of an immunoBody™ construct when delivered as a DNA vaccine.
The CDRL1 site was replaced with the I-Ab restricted helper epitope from the Hep B surface antigen, the I-Ad restricted helper epitope from the influenza haemagglutinin or the human HLA-DR4 epitope from the melanoma associated antigen gp100 (). C57Bl/6, Balb/c or HLA-DR4 transgenic mice were immunized three times at weekly intervals with ImmunoBody™ DNA via the gene gun. Splenocytes were subsequently analyzed for the presence of epitope specific responses ex vivo in IFNγ elispot assay. All three epitopes were processed and stimulated significantly higher frequency T cell responses in C57Bl/6, Balb/c or HLA-DR4 transgenic mice compared to control (p < 0.0001, p < 0.0001 and p = 0.0004 respectively; ).
The CDRL3 site was also substituted with the I-Ab restricted helper epitope from the HepB surface antigen, the human HLA-DR4 epitopes from the melanoma associated antigens gp100, tryrosinase or the HLA-DR1 restricted epitope from the melanoma antigen Triosephosphate isomerize (TPI) (). C57Bl/6, HLA-DR4 transgenic or HLA-DR1 transgenic mice were immunized with ImmunoBody™ DNA via gene gun. All three epitopes from melanoma associated antigens stimulated high frequency T cell responses in HLA-DR4 and HLA-DR1 transgenic mice compared to control (p = 0.0005, p = 0.05 and p = 0.05 respectively; ).
ImmunoBody™ vaccines can therefore stimulate helper responses to a wide range of epitopes encoded within a range of heavy and light chain CDRs. To see if a similar wide diversity of CTL responses could be generated a range of CTL epitopes were encoded within a range of CDRs and the constructs screened for their ability to stimulate CTL responses.
CTL responses can be generated from a wide range of different epitopes.
Although secretion of intact antibody was not required for stimulation of CTL responses to the TRP-2 epitope, it was unclear if this could be repeated with other CTL epitopes. The CDRH1 site was therefore replaced with HLA-A2 restricted epitopes from the tumor associated antigen gp100, telomerase, Tie-2 or MAGE-3 (). ImmunoBody™ constructs containing the heteroclitic gp100 210M epitope and the heteroclitic Tie-2 epitope (Z84) stimulated high frequency T cell responses in HLA-A2 transgenic mice compared to control (p < 0.0001 and p = 0.02 respectively) (). However specific responses were unable to be generated for the telomerase, wild type gp100 and MAGE-3 epitopes.
The CDRH2 site was also substituted with the H-2Kb restricted epitope from ovalbumin, the H-2Kb/HLA-A2 restricted epitope from the melanoma-associated antigen TRP-2, two telomerase HLA-A2 restricted epitopes or the H-2Kd restricted epitope from the Hep B surface antigen (). Mice immunized with ImmunoBody™ constructs containing the TRP-2 epitope stimulated high frequency T cell responses in both C57Bl/6 and HLA-A2 transgenic mice compared to control (p < 0.0001) (). ImmunoBody™ construct containing the Hep B epitope stimulated significant epitope specific T cell responses in Balb/c mice (p < 0.0001), and the ovalbumin epitope stimulated high frequency epitope specific responses in C57Bl/6 mice (p < 0.0001) (); however, the telomerase epitope again failed to stimulate any response.
The CDRH3 site was also replaced with the H-2Kb/HLA-A2 restricted epitope from TRP-2 or wild type and heteroclitic HLA-A2 restricted epitopes from VEGFR2 (). The TRP-2 epitope was processed and presented from the ImmunoBody™ construct to generate epitope specific T cell responses in C57Bl/6 mice compared to control (p < 0.0001), but ImmunoBody™ constructs containing the VEGF epitopes failed to stimulate any response in HLA-A2 transgenic mice ().
In order to confirm that responses were mediated by CD8 cells, CD8 positive cells were depleted prior to analysis in IFNγ elispot assay from mice immunized with a construct containing H-2Kb restricted TRP2 epitope in CDRH2 and HepB helper epitope in CDRL1. Depletion of CD8 T cells lead to abolition of the TRP2 specific responses (p < 0.05), but did not affect the HepB helper peptide specific responses, suggesting that the TRP2 specific responses is mediated by CD8 T cells (). These results suggested that ImmunoBody™ could stimulate CTL response to some, but not all, inserted epitopes. To assess the possibility of stimulating multiple CTL responses, several CTL epitopes were engineered into the same construct.
Multiple CTL responses can be generated from different epitopes within the same immunoBody™ construct.
To avoid potential outgrowth of antigen loss variants following vaccination against a single antigen, multiple epitopes from different antigens were cloned within different CDRs. The HLA-A2 restricted heteroclitic gp100 210M epitope was engineered into the CDR H1 site alongside the HLA-A2 restricted TRP2 epitope in the CDR H2 site of the same construct. The HepB CD4 epitope was present in the CDR L1 site. HLA-A2 transgenic mice were immunized three times at weekly intervals by intradermal immunization with ImmunoBody™ DNA via the gene gun. Splenocytes were subsequently analyzed by IFNγ elispot for the presence of epitope specific CD8 and CD4 responses. shows that significant responses are generated to the gp100 210M and TRP2 CD8 epitopes as well as the HepB CD4 epitope (p < 0.0001). To assess if the cellular immune responses generated by ImmunoBody™ were strong enough to inhibit tumor growth, an ImmunoBody™ expressing the TRP-2 CTL epitope and the HLA-DR4 gp100 helper epitope were used to immunize C57BL mice who were subsequently challenged with B16 tumor.
ImmunoBody™ immunization prevents tumor growth.
ImmunoBody™ DNA immunization is capable of generating high frequency CD8 and CD4 epitope specific T cell responses. Evidence suggests that the presence of antigen specific CD4 help alters the chemokine gradient and enhances extravasation of T cells into the tumor site. Therefore, the tumor specific H-2Kb restricted TRP2 CD8 epitope and HLA-DR4 restricted gp100 CD4 epitope were engineered into the CDRH2 and CDRH3 sites, respectively. This construct was analyzed for its ability to prevent the growth of the aggressive B16F1 melanoma line in vivo. HLA-DR4 transgenic mice were immunized with ImmunoBody™ DNA three times at weekly intervals, and then they were challenged with B16F1 tumor cells. The results () demonstrated that mice immunized with ImmunoBody™ DNA vaccine have significantly slower tumor growth at day 22 post tumor implant (p = 0.0016) compared to animals immunized with control ImmunoBody™ DNA containing no T cell epitopes.
Discussion
Several groups have used antibodies as vectors, replacing CDRH3 with helper and B cell epitopes which stimulated immune responses.Citation23,Citation24,Citation37–Citation39 Zaghouani et al. also attempted to replace CDRH3 with class I restricted CTL epitopes. Although they showed that transfectomas expressing recombinant Ig encompassing an MHC class I CTL epitope from the nucleoprotein of influenza virus (NP-Ig) were capable of inducing CTL responses, the purified Ig was unable to do so.Citation40,Citation41 It was concluded that FcyR initiated processing pathways do not result in presentation of T cell epitopes via MHC class I due to the lack of intersection with the endogenous pathway of processing and presentation. More recent studies linking T cell epitopes to a Fab targeting the high affinity receptor FcγR1,Citation42 our own studies with a human anti-idiotypic antibody expressing T cell mimotopesCitation5,Citation20,Citation21 or using antigen/antibody immune complexes suggest otherwise, and have pinpointed the roles of various FcγRs in the process.Citation43,Citation44
Typically, exogenous antigens are presented on class II MHC and internal antigens are processed on class I MHC. Thus antigen presenting cell phagocytosis of an exogenous antigen would normally lead to presentation of the antigen on class II MHC leading to activation of CD4 helper cells. However, it has recently been shown that it is possible to cross present exogenous antigen on MHC class I and that endogenous antigens are often cross presented on MHC class II by autophagy.Citation45,Citation46 One route to allow cross presentation of exogenous antigens on MHC class I is via FcγRs. We and others have shown that the high affinity FcγRI, allows efficient cross presentation of monomeric human IgG1 or mouse IgG2a.Citation19,Citation47 We have shown that a human monoclonal IgG1 anti-idiotypic antibody, 105AD7, can stimulate helper and cytotoxic T cell responses in over 300 cancer patients with no associated toxicity.Citation5–Citation21 If the Fc region of this antibody is removed it is 1,000 fold less efficient at stimulating T cells.Citation18 Similarly, an anti-idiotypic antibody mimicking CEA was very inefficient at stimulating human T cells as a mouse IgG2b, but, when chimerized to a human IgG1, the antibody stimulated CTLs that killed tumor cells expressing CEA.Citation19 Immature circulating DCs in the blood express only low levels of FcγRI to avoid binding serum Ig; its expression is transiently upregulated by IFNγ on extravasation into inflamed tissue.Citation48,Citation49 It can then bind, internalize and process any IgG whether free or forming small immune complexes within the inflamed tissue. FcγRI is then downregulated, and the activated DCs migrate to the local draining lymph node and present antigen. In contrast, larger immune complexes can be cross presented by FcγRIIa (FcγRIV in mice), but only if the inhibitory FcγRIIb receptor is blocked or downregulated.Citation50 Recent studies with the mouse IgG2b expressing the NP CTL epitope (NP-Ig) have shown that it is possible to stimulate CTL responses to this antibody if it is co-administered with the TLR agonist dsRNA which upregulates FcγRIV and downregulates FcγRIIb.Citation51 In contrast, we have shown that immunising with a DNA vaccine incorporating CTL and helper epitopes within a human IgG1 or mouse IgG2a framework without any additional adjuvants stimulates strong responses to a wide range of epitopes.
ImmunoBody™ is an antibody vector system which allows rapid insertion of T cell epitopes into a variable region that can be exchanged between isotypes. This has been achieved by developing human IgG1/murine IgG2a double expression vectors with 1–6 CDRs replaced with restriction endonuclease sites. This allows quick insertion of any oligonucleotides expressing single or multiple epitopes. In this report we have shown that it is possible to express CTL epitopes from a range of viral (Hep B surface antigen), foreign (ovalbumin), self (Tie-2) and tumor associated antigens (TRP-2, gp100) within the variable region of a human IgG1 or mouse IgG2a antibody and use the DNA that encodes these antibodies as a vaccine to stimulate high frequency T cell responses in C57BL, Balb/c, HLA-A*0201 transgenic and HLA-DR4 transgenic mice. Responses observed in elispot assays are not believed to be due to the effect of natural killer (NK) cells, as depletion of CD8 T cell subset abrogated epitope specific responses. In addition to this, IFNγ release was only observed with specific epitope peptides and not control peptides, which would not be the case if responses were a result of NK cell activity. It was also possible to stimulate helper T cell responses to epitopes from viral (Hep B, Influenza nucleoprotein) or melanoma associated antigens (gp100, TPI and tyrosinase). As expected, the highest frequency responses were generated to the foreign and viral epitopes. Three immunizations were required to stimulate optimal T cell responses that convert to memory cells and can be efficiently reactivated.
Comparison of the ImmunoBody™ DNA vaccine encoding TRP-2 epitope to whole murine TRP2 antigen DNA immunization revealed significantly enhanced responses from ImmunoBody™ DNA. Previous studies have shown that xenogenic DNA immunization can stimulate a response to TRP-2, but breaking tolerance to this epitope from syngeneic DNA has been largely unsuccessful.Citation52 The generation of anti-tumor immunity to a syngeneic self antigen has been demonstrated, but only when enhanced by adjuvants and in the absence of regulatory T cells.Citation53–Citation55 This would suggest that the presence of regulatory determinants within syngeneic whole tumor antigen sequences that are absent in xenogeneic sequence prevents the generation of efficient T cell responses. The removal of immunogenic epitope sequences out of the regulatory environment of the whole antigen and into an inert carrier such as an ImmunoBody™ therefore enhances the immune responses. Furthermore, responses to the melanoma associated antigen TRP2 from ImmunoBody™ DNA vaccine were strong enough to show in vivo growth delay of the aggressive B16F1 melanoma tumor.
Also of interest was that all of the epitopes shown to be presented by the immunoproteasome were efficiently processed and presented, but epitopes that are destroyed by the immunoproteasome (gp100 wild type) or peptide epitopes that have never been shown to be presented as immunogens within whole proteins (MAGE3, TERT and VEGFR2) all failed to elicit responses. The TERT epitopes were placed in several CDRs to determine whether adjacent residues within the antibody were altering processing, but there was still no response. The VEGFR2 and MAGE-3 epitopes were incorporated with adjacent residues from their antigens or with poly alanines to facilitate processing, but none of these interventions resulted in the efficient generation of responses (data not shown). Future studies will confirm if ImmunoBody™ preferentially targets activated DCs expressing the immunoproteasome, and if only epitopes liberated by these proteosomes will be suitable candidates for ImmunoBody™ targets.
As anticipated, replacement of CDR regions other than CDR3 with any of the epitopes prevented heavy and light chain association and resulted in predominantly heavy chain secretion. However, contrary to expectation, DNA constructs encoding antibody that showed no or very low levels of secretion of intact antibody gave as good or better responses than constructs expressing intact antibody. Initially it was believed that gene gun immunization functioned via the direct transfection of APCs that travel to lymph nodes where they activate T cells.Citation56–Citation58 However, more recent studies have suggested that cross presentation is the major route for CTL induction following this type of immunization.Citation59 The apparent difference in the frequency of responses generated from ImmunoBody™ DNA compared to the protein equivalent suggests that the direct transfection of skin APCs does play a role in the generation of these immune responses. There is evidence in the literature that peptide epitopes can commonly be generated from defective polypeptides that have errors in translation or post-translational modification, or are misfolded. Collectively, these are known as defective ribosomal products (DRiPs).Citation60–Citation62 It is therefore possible that insertion of epitopes into CDRs that disrupt antibody folding enhance the processing of antigenic peptides from ImmunoBody™ DNA. However, this is not the only method by which ImmunoBody™ DNA induces responses, as abrogation of protein secretion via removal of the leader sequence led to reduction in responses, suggesting that secreted protein is also necessary for induction of high frequency responses. Our study therefore suggests that the high frequency CTL responses stimulated by ImmunoBody™ DNA vaccines are a result of both direct and cross presentation. In combination with evidence from our studies with a human anti-idiotypic antibody, this would suggest that the route of cross presentation may be through FcγR targeting of antigen presenting cells.Citation18,Citation19 Further studies on Fc knockout mice will be required to confirm this hypothesis.
Although only low levels of intact antibody were detected from some of the most efficient ImmunoBody™ constructs, these may be important in stimulating the immune response, as several studies have shown that immunization with low levels of peptides stimulate high avidity T cell responses.Citation63 The lack of a requirement for large amounts of intact antibody by the DNA vaccine has a significant advantage as it allows greater flexibility in terms of the number and length of epitopes that can be incorporated. It may be necessary to incorporate epitopes from several tumor associated antigens to prevent selection of antigen loss variants, and it may also be necessary to incorporate several epitopes binding to a range of MHC class I alleles to allow the vaccine to be used in a wide range of patients. In contrast, incorporation of epitopes within the CDRH3 to allow protein folding is restricted by the hydrophilicity of the inserted sequence. Our studies show that CDRH3 can be substituted with linked multiple epitopes of up to 29 amino acids in length, as long as the residue substitutions preserve the hydropathic profile of the wild type sequence. This is less of a problem for helper epitopes, and indeed we could successfully incorporate two helper epitopes (29 amino acids) within the CDRH3 and still get intact protein. CTL epitopes are more hydrophobic as they bind tightly within the groove of a MHC molecule. Although it was possible to incorporate a single CTL epitope within CDRH3 or CDRL3, two linked CTL epitopes failed to allow secretion of substantial amounts of intact antibody.
It is believed that the presence of CD4 help is required for the generation of efficient CD8 memory responses.Citation66,Citation67 The IgG1 human constant region may provide foreign linked help in the mice models as the mouse IgG2a constructs which would lack this help, gave lower frequency responses. This problem was overcome by incorporating linked help within the heavy chain of the mouse IgG2a construct. An ideal vaccine may include incorporation of helper epitope within the heavy chain to allow efficient priming of CTLs and an antigen specific helper epitope within the light chain, which would allow CD4 amplification of the immune response at the tumor site.Citation68,Citation69
In conclusion, we have designed a robust and efficient DNA vector that encodes a human IgG1 antibody with its CDRs replaced with restriction sites to enable rapid insertion of single or multiple T cell epitopes. These DNA vaccines efficiently present a wide range of CTL and helper epitopes and stimulate high frequency CTL and helper responses. DNA vaccines are quicker and cheaper to manufacture and stimulate strong T cell responses in animal models. Early studies to translate them into humans have shown limited responses; however, this has recently been significantly improved by the development of enhanced delivery devices.Citation64,Citation65 This technology is a novel approach to vaccination and demonstrates the potential for the system to be used as a multivalent vaccine for many cancer types and infectious disease.
Materials and Methods
Generation of the ImmunoBody™ single heavy and light chain vectors.
Multi step cloning was required in order to generate the ImmunoBody™ heavy and light chain plasmids pOrigHIB and pOrigLIB ( and B). These contained the murine variable heavy (VH) and kappa (VL) regions PCR cloned from a DeImmunized mouse human chimeric antibody (SC100) and inserted upstream and in frame with the human kappa and heavy IgG1 constant regions. Each chain was under the control of the CMV early promoter and BGH polyadenylation signal.
Replacement of CDRs with unique restriction recognition sequences.
The CDR regions were removed and replaced with unique restriction sites by overlap extension PCR using the heavy chain variable region oligonucleotides H1, H2, H3 and huHeClonR IgG constant region reverse primer () for first round PCR. Similarly, for the light variable region, the oligonucleotides L1, L2, L3 and reverse primer huLiClonR were designed to replace each of the three CDRs (). Resulting PCR product was then used in a subsequent PCR as a reverse primer in conjunction with the CMV forward primer. The amplified DNA fragment was cloned directly into the TA TOPO vector pCR2.1 (Invitrogen, USA) and clones sequenced to confirm amplification of the VH and VL region devoid of the CDRs and replacement of restriction site. The different versions were then cloned back into pOrigHIB and pOrigLIB using HindIII/AfeI and BamHI/BsiWI with direct replacement of the parental wild type DeImmunized SC100 VH and VL regions ().
Construction of the ImmunoBody™ double expression vector.
To generate the ImmunoBody™ double expression vector pDCOrig, pOrigHIB was linearized and the entire light chain expression cassette consisting of the CMV promoter, antibody light chain and the BGH polyA signal were inserted to form the construct pDCOrig (). Orientation of the light chain cassette within pDCOrig was confirmed by restriction analysis.
Generation of mouse IgG2a pDCOrig IB15.
For amplification of the murine IgG2a constant region cDNA was used as a template, synthesized from total RNA isolated from the hybridoma cell line 337, with the forward primer moIgG2aC1AfeFor containing the restriction site Afe1 and the reverse primer moIgG2aXbaRev harboring a XbaI site after the stop codon. The murine IgG2a constant region was cloned in frame with the murine VH region into the Afe1/XbaI sites of the vector pOrigHIB effectively replacing human IgG1 generating the single chain vector pMoOrigHIB. A section of pMoOrigHIB containing the murine IgG2a constant region was transferred from the single construct into the double expression vector containing, gp100 210M epitope in CDRH1, TRP2 epitope in CDRH2 and HepB helper epitope in CDRL1, in frame with the murine VH region using AfeI and the single cutter AvrII located in the SV40 promoter to generate a intermediate double expression vector still containing a human kappa region.
For amplification of the murine kappa region the cDNA was used as a template with the primers moLC1BsiFor containing a BsiWI site and moLCXhoIRev containing an XhoI site after the stop codon (). The murine kappa region was excised and ligated into the BsiWI/XhoI sites of the vector pOrigLIB HepB help/L1 replacing the human kappa constant generating the intermediate vector pMoLIBBsi HepB help/L1. The BsiWI site was removed to retain wild type murine kappa sequence. This was achieved by overlapping PCR with reamplification of the murine full length chain. The full length murine kappa chain containing HepB help in the L1 site was excised and cloned into the BamHI/XhoI sites of the intermediate double expression vector described above in direct replacement of the DeImmunized human kappa chain to generate the murine IgG2a construct with gp100 210M epitope in CDRH1, TRP2 epitope in CDRH2 and HepB helper epitope in CDRL1. Sequence was confirmed.
The different versions of the VL regions containing L1, L2 and L3 sites singly and in combination were inserted into the intermediate single chain vector pMoLIBBsi HepB help/L1using BamHI/BsiWI and the above method of overlapping extension PCR adopted utilizing the same primers to generate the murine full length single chain vectors pMoOrigLIB containing the L1, L2 and L3 sites. After insertion of epitopes into these sites within these vectors the murine VL region can be effectively exchanged into the double murine IgG2a expression vector using BamHI and ClaI.
Transfection.
Ten microliters of Lipofectamine 2000 (Invitrogen) and 4 µg of plasmid DNA were diluted with Optimem I Reduced Serum medium (Gibco BRL, USA), gently mixed and incubated at room temperature for 20 minutes to form complexes. The complexes were added to CHO cells (Chinese hamster ovary cells, ECACC, UK) in 35 mm wells of a 6-well tissue culture plate. After 24 hours incubation at 37°C, in 5% CO2 cells were harvested and plated into 96 well tissue culture plates in medium containing Zeocin at a final concentration of 300 µg/ml (Invivogen, USA). Resistant clones were cloned for Ig secretion by capture ELISA.
Sandwich ELISA.
Falcon 96-well flexible plates were coated, overnight at 4°C, with 50 µl of anti-human IgG, Fc specific antibody (Sigma, UK) or anti-human kappa light chain antibody (Dako, Denmark) at 10 µg/ml in PBS. Plates were washed three times with 200 µl/well PBS-Tween 20 (0.05%) and wells blocked with 1% fish skin gelatin (Sigma) in PBS (1% FSG/PBS). Plates were incubated 1 hr at room temperature and washed with PBS-Tween 20 (0.05%). Tissue culture supernatant containing expressed ImmunoBody™ or purified ImmunoBody™ protein (50 µl) was added to the wells, in triplicate, and plates were incubated for 1 hr at room temperature. Plates were washed with PBS-Tween 20 (0.05%) and bound ImmunoBody™ was detected by adding 50 µl/well of peroxidase-conjugated anti-human IgG, Fc specific antibody (Sigma) or anti-human kappa light chain antibody (Sigma), diluted 1/2000 in 1% FSG/PBS, and incubated 1 hr at room temperature. Plates were washed with PBS-Tween 20 (0.05%) and developed by adding TMB substrate (R&D Systems, USA) at 50 µl/well. Absorbance was measured at 650 nm.
SDS-PAGE and western analysis.
To check for formation of intact antibody, purified protein and supernatants containing protein secreted by transfected CHO-S cells with the ImmunoBody™ constructs were ran on a 12% SDS-PAGE under non reducing conditions. Western blot analysis was performed according to Towbin et al.Citation36 Purified antibodies (2 µg/Lane) and supernatant from CHO-S cells transfected with the ImmunoBody™ constructs were electrophoresed on a 12% SDS-PAGE gel and then blotted onto 0.45 µm nitrocellulose membrane (Hi-bond, GE Healthcare, USA). The membrane was blocked by soaking in 1% BSA in PBS-Tween 20 (0.1%) overnight at room temperature. After washing in PBS-Tween 20 (0.1%) the Horseradish peroxidize (HP)-conjugated goat α-human IgG specific (Sigma; 1:2000 dilution) and HP-conjugated α human kappa light chains (Sigma; 1:2000 dilution) antibodies in 1% BSA/PBS-Tween 20 were added for 1 hour at room temperature. After washing, the blot was developed using ECL reagents (GE Healthcare) according to manufacturer's instructions and exposed to photographic film.
Mice and immunizations.
Animal work was carried out under a Home Office approved project licence. Female C57Bl/6 (Charles River, UK), Balb/c (Harlan, UK), HLA-DR4 transgenic (Taconic, USA) or HLA-A2 transgenic (HHDII; Pasteur Institute, Paris) were used between 6 and 12 weeks of age. DNA was coated onto 1.0 µm gold particles (BioRad, Hemel Hempstead, UK) using the manufacturer's instructions and administered intradermally by the Helios Gene Gun (BioRad). Each mouse received 1 µg DNA/immunization. For protein immunizations 100 µg of protein in complete Freund's adjuvant was administered s.c. for the prime and 100 µg of protein in incomplete Freund's adjuvant for booster immunizations. Mice were immunized at 0, 7 and 14 days and spleens removed at day 20 for analysis unless stated otherwise. For tumor challenge experiments mice were immunized as above and concurrent with the final immunization injected with 2.5 × 104 B16F1 cells subcutaneously. Tumor growth was monitored at 3–4 day intervals and mice were humanely euthanized once tumor reached >10 mm in diameter.
Ex vivo elispot assay.
Elispot assays were performed using murine IFNγ capture and detection reagents according to the manufacturer's instructions (Mabtech, Sweden). In brief, anti-IFNγ antibodies were coated onto wells of 96-well Immobilin-P plate and triplicate wells were seeded with 5 × 105 splenocytes which were harvested by spleen perfusion, washed and resuspended in RPMI 1640 10% foetal calf serum. Synthetic peptides, at a variety of concentrations, were added to these wells in triplicate and incubated for 40 hrs at 37°C. After incubation, captured IFNγ was detected with by a biotinylated anti-IFNγ antibody and development with a streptavidin alkaline phosphatase and chromogenic substrate. Spots were analyzed and counted using an automated plate reader.
Statistical analysis.
Comparative analysis of the Elispot results was performed by applying the t-test with values of p calculated accordingly.
Figures and Tables
Figure 1 Construction of the ImmunoBody™ double expression vector pDCOrig. (A) Heavy chain vector pOrigHIB. The wild type DeImmunised VH region of antibody SC100 was cloned using HindIII/AfeI in frame with the human IgG1 Fc constant region. The Fc region comprises the CH1, CH2, CH3 domains and the hinge region. High-level expression in mammalian cells is driven from the CMV immediate early promoter. BGH polyadenylation signals downstream of the OrigHIB human IgG1 chain to ensure mRNA stability and effective termination. EM7 is a bacterial promoter that controls expression of the zeocin resistance gene allowing antibiotic selection in E. coli while the SV40 early promoter upstream of the resistance gene allows selection in mammalian cells. SV40 polyadenylation signals downstream of the resistance gene in order to direct proper processing of the 3′end of the zeor mRNA. The vector also contains within its backbone the ColE1 origin of replication for propagation in bacteria. (B) Light chain vector pOrigLIB. The wild type deimmunized VL region of antibody SC100 was cloned using BamHI/BsiWI in frame with the human kappa constant region. High-level expression in mammalian cells is driven from the CMV immediate early promoter. BGH polyadenylation signals downstream of the OrigLIB chain to ensure mRNA stability and effective termination. The vector also includes the ColE1 origin of replication and the antibiotic resistance gene for ampicillin allowing propagation and selection in bacteria. (C) Double expression vector pDCOrig. Once all epitopes have been incorporated into the VH and VL sites within the single vectors they are transferred into the double expression vector utilizing as highlighted HindIII/AfeI and BamHI/BsiWI in frame with their respective human constant regions. The Fc region of the heavy chain comprises of the CH1, CH2, CH3 domains and the hinge region. High-level expression of both the heavy and light chains in mammalian cells is driven from the CMV immediate early promoter. (D) Complimentary determining DNA sequences were removed by overlapping PCR and exchanged for the unique restriction sites RE1, RE2 and RE3 (EcoRV, SspI and HpaI) within the single light chain and heavy chain constructs RE4, RE5 and RE6 (FspI, MscI and SrfI respectively) singly and in combination.
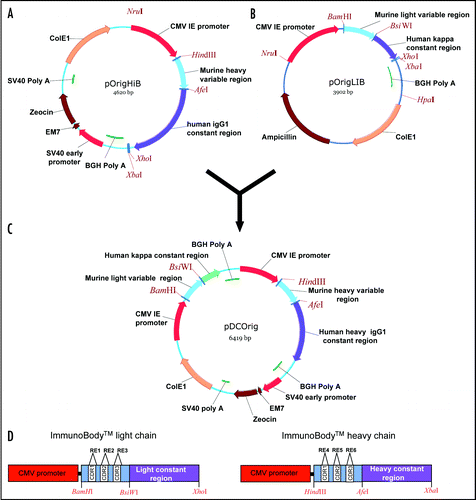
Figure 2 Antibody secretion. (A) A total of 2 µg of purified protein (lanes 1, 3 and 5) and supernatant from transfected CHO-S cells (lanes 2, 4 and 6) were loaded onto a 12% SDS-PAGE gel and subjected to electrophoresis under non-reducing conditions. Wild type ImmunoBody™ antibody is shown in lanes 1 and 2, TRP2 grafted into the CDRH2 site alongside the gp100 210M CTL in CDRH1 and HepB help CD4 epitope in CDRL1 in lanes 3 and 4 and the TRP2 CTL epitope in CDRH3 in lanes 5 and 6. The nitrocellulose blot was incubated with a HRP goat anti human IgG Fc specific antibody. (B) Western blot analysis was carried out as above however the nitrocellulose blot was incubated with a HRP anti human kappa light chain antibody. (C) Hydropathicity plots of CDRH3 within the wild type ImmunoBody™ heavy chain and those incorporating CTL (TRP2) and CD4 (HepB help) epitopes. The Kyte and DoolitleCitation70 hydropathicity index was utilized to calculate the hydropathicity distribution.
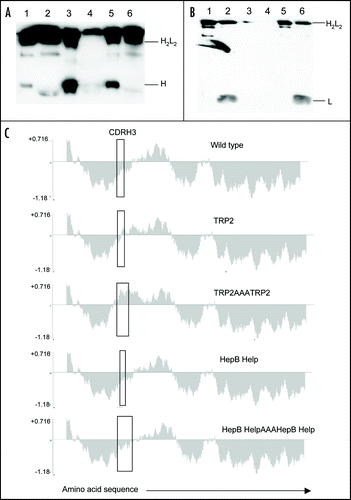
Figure 3 ImmunoBody™ DNA immunization. (A) C57Bl/6 mice were immunized at days 0, 7 and 14 with an ImmunoBody™ protein containing the H-2Kb epitope in the CDRH3 site (n = 6) or the equivalent DNA (intact) (n = 12) or ImmunoBody™ DNA containing the H-2Kb restricted TRP2 epitope in CDRH2 (non associated; n = 9). On day 19 splenocytes from immunized mice were analyzed ex vivo for the presence of epitope specific responses. Responses are measured as spots/million splenocytes. (B) C57Bl/6 mice were immunized on days 0, 7 and 14 with an ImmunoBody™ DNA construct containing TRP2 epitope in CDRH2 with or without leader sequence. On day 19 splenocytes were analyzed by IFNγ elispot assay against TRP2 peptide in triplicate. Responses are measured as spots/million splenocytes and normalised against an irrelevant peptide control (n = 4). (C) C57Bl/6 mice were immunized at days 0, 7 and 14 with an ImmunoBody™ DNA containing the H-2Kb epitope in the CDRH2 site (n = 6) or whole murine TRP2 antigen in pcDNA3 (n = 6) or pOrig vectors (n = 6). On day 19 splenocytes from immunized mice were analyzed ex vivo for the presence of epitope specific responses. Responses are measured as spots/million splenocytes. (D) C57Bl/6 mice were immunized with human IgG1 DNA containing TRP2 epitope in CDRH2 at days 0 and 7 (n = 6), or 0, 7 and 14 (n = 6). Splenocytes were analyzed on day 19 for the presence of TRP2 epitope specific responses by IFNγ elispot. Responses are measured as spots/million splenocytes. (E) C57Bl/6 mice were immunized with ImmunoBody™ DNA at days 0, 7 and 14 via gene gun (n = 6). Responses were analyzed at day 20, day 48 post primary immunization. A parallel group of mice was boosted at day 42 (n = 6). Responses were tested in ex vivo elispot assay against TRP2 peptide and an irrelevant control in triplicate. Responses are measured as spots/million splenocytes. All results are an average of two independent experiments.
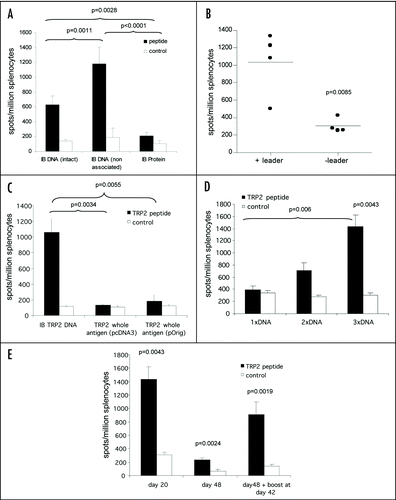
Figure 4 Linked help is required for optimal CTL responses. (A) C57Bl/6 or HLA-A*0201 transgenic mice were immunized at day 0, 7 and 14 with ImmunoBody™ heavy chain only DNA constructs containing the H-2Kb restricted TRP2 epitope in CDRH2 or the HLA-A*0201 restricted gp100 210M epitope in CDRH1. On day 19, splenocytes were analyzed by IFNγ elispot assay against gp100 210M peptide, TRP2 peptide and control. Responses are measured as spots/million splenocytes (n = 6). (B) C57Bl/6 mice were immunized at days 0, 7 and 14 with an ImmunoBody™ DNA containing the H-2Kb restricted TRP2 epitope in CDRH2, the HLA-A*0201 restricted epitope gp100 210M in CDRH1 and the I-Ab restricted HepB helper epitope in CDRL1 with either human IgG1 or murine IgG2a constant domains. On day 19, splenocytes were analyzed by IFNγ elispot assay against TRP2 peptide, HepB helper peptide and control. Responses are measured as spots/million splenocytes (n = 6). (C) HLA-DR*0401 transgenic mice were immunized at days 0, 7 and 14 with an ImmunoBody™ DNA containing the gp100 DR4 epitope in CDR H1, TRP2 epitope in CDR H2 and gp100 DR7 epitope in CDR H3 with either human IgG1 or murine IgG2a constant domains. On day 19, splenocytes were analyzed by IFNγ elispot assay against TRP2 peptide, gp100 DR4 helper peptide and control. Responses are measured as spots/million splenocytes (n = 6). (D) C57Bl/6 (n = 48), Balb/c (n = 9) or HLA-DR*0401 (n = 6) transgenic mice were immunized on days 0, 7 and 14 with an ImmunoBody™ construct containing either I-Ab restricted HepB, I-Ad restricted Influenza or HLA-DR*0401 restricted gp100 epitopes in CDR L1. On day 19 splenocytes were analyzed by IFNγ elispot assay against HepB, Influenza or gp100 helper peptides and an irrelevant control. Responses are measured as spots/million splenocytes. (E) C57Bl/6 (n = 6), HLA-DR*0101 (n = 6) or HLA-DR*0401 (n = 6) transgenic mice were immunized on days 0, 7 and 14 with an ImmunoBody™ construct containing either I-Ab restricted HepB or HLA-DR*0401 restricted gp100 or tyrosinase or HLADR* 0101 restricted triosephosphate isomerase (TPI) epitopes in CDR L3. On day 19 splenocytes were analyzed by IFNγ elispot assay against HepB, gp100, TPI or tyrosinase helper peptides and an irrelevant control. Responses are measured as spots/million splenocytes. All results are an average of at least two independent experiments.
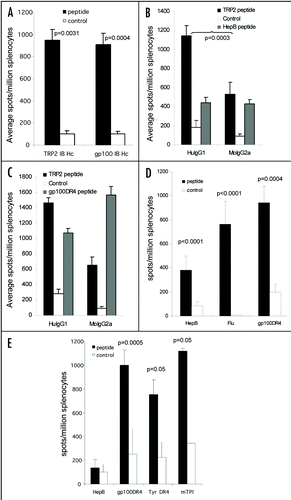
Figure 5 CTL epitopes can be processed and presented to elicit high frequency responses. (A) HLA-A2 transgenic mice were immunized on days 0, 7 and 14 with an ImmunoBody™ construct containing either HLA-A*0201 restricted gp100 210M (n = 12), gp100 wild type (n = 6), Mage3 (n = 6), hTERT (n = 6) or Tie2 (n = 6) epitopes in CDRH1. On day 19 splenocytes were analyzed by IFNγ elispot assay against gp100 210M, gp100 wild type, Mage3, hTERT or Tie2 peptides and an irrelevant control. Responses are measured as spots/million splenocytes. (B) C57Bl/6, Balb/c or HLA-A*0201 transgenic mice were immunized on days 0, 7 and 14 with an ImmunoBody™ construct containing the H-2Kb restricted TRP2 epitope (n = 50) which is also restricted through HLA-A*0201 (n = 12), the H-2Kd restricted HepB epitope (n = 6), the H-2Kb Ovalbumin epitope (n = 6) or the HLA-A*0201 restricted hTERT epitope (n = 6) in CDRH2. On day 19 splenocytes were assayed ex vivo in IFNγ elispot assay against relevant and control peptides. Responses are measured as spots/million splenocytes. (C) C57Bl/6 or HLA-A*0201 transgenic mice were immunized on days 0, 7 and 14 with an ImmunoBody™ construct containing the H-2Kb restricted TRP2 epitope (n = 12) or HLA-A*0201 restricted VEGFR2 epitopes (n = 6) in CDRH3. On day 19 splenocytes were assayed for presence of specific responses by IFNγ elispot assay. Responses are measured as spots/million splenocytes. (D) Splenocytes from immunized mice were depleted of CD8 T cells and analyzed against TRP2 peptide, HepB helper peptide and a media control in triplicate for the presence epitope specific responses in IFNγ elispot assay in triplicate. Responses are measured as spots/million splenocytes (n = 4). (E) HLA-A*0201 transgenic mice were immunized on days 0, 7 and 14 with an ImmunoBody™ construct containing HLA-A*0201 restricted TRP2 epitope in CDRH2, the HLA-A*0201 restricted gp100 210M epitope in CDRH1 and the I-Ab restricted HepB helper epitope in CDRL1. On day 19 splenocytes were analyzed by IFNγ elispot assay against relevant peptides and an irrelevant control in triplicate. Responses are measured as spots/million splenocytes (n = 12). All results are an average of at least two independent experiments.
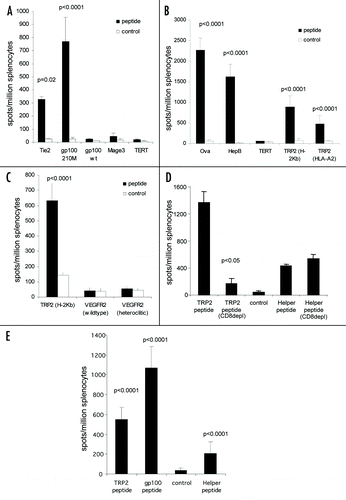
Figure 6 Immune responses generated by ImmunoBody™ DNA vaccination can delay tumour growth. HLA-DR*0401 transgenic mice were immunised with ImmunoBody™ DNA containing the H-2Kb restricted TRP2 epitope in CDRH2 and the HLA-DR*0401 restricted gp100 epitope in CDRH3 via gene gun at days 0, 7 and 14. On day 14, mice were injected s.c. with 2.5 × 104 B16F1 tumour cells. Tumour growth was monitored at 3–4 day intervals using a calliper and displayed as tumour volume at day 22 post tumour implant (n = 10).
Table 2 List of CDR replacement enzymes and epitope oligonucleotides sequences

Table 1 Primers used in this study
Table 3 CTL and helper epitopes
Table 4 ImmunoBody™ protein secretion
References
- Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY. Optimizing engagement of the immune system by anti-tumor antibodies: an engineer's perspective. Drug Discov Today 2007; 12:898 - 910
- Selenko N, Majdic O, Jager U, Sillaber C, Stockl J, Knapp W. Cross-priming of cytotoxic T cells promoted by apoptosis-inducing tumor cell reactive antibodies?. J Clin Immunol 2002; 22:124 - 130
- de Cerio AL, Zabalegui N, Rodriguez-Calvillo M, Inoges S, Bendandi M. Anti-idiotype antibodies in cancer treatment. Oncogene 2007; 26:3594 - 3602
- Sharav T, Wiesmuller KH, Walden P. Mimotope vaccines for cancer immunotherapy. Vaccine 2007; 25:3032 - 3037
- Pritchard-Jones K, Spendlove I, Wilton C, Whelan J, Weeden S, Lewis I, et al. Immune responses to the 105AD7 human anti-idiotypic vaccine after intensive chemotherapy, for osteosarcoma. Br J Cancer 2005; 92:1358 - 1365
- Maxwell-Armstrong CA, Durrant LG, Buckley TJ, Scholefield JH, Robins RA, Fielding K, et al. Randomized double-blind phase II survival study comparing immunization with the anti-idiotypic monoclonal antibody 105AD7 against placebo in advanced colorectal cancer. Br J Cancer 2001; 84:1443 - 1446
- Austin EB, Robins RA, Durrant LG, Price MR, Baldwin RW. Human monoclonal anti-idiotypic antibody to the tumour-associated antibody 791T/36. Immunol 1989; 67:525 - 530
- Austin EB, Robins RA, Baldwin RW, Durrant LG. Induction of delayed hypersensitivity to human tumor cells with a human monoclonal anti-idiotypic antibody. J Natl Cancer Inst 1991; 83:1245 - 1248
- Robins RA, Denton GW, Hardcastle JD, Austin EB, Baldwin RW, Durrant LG. Antitumor immune response and interleukin 2 production induced in colorectal cancer patients by immunization with human monoclonal anti-idiotypic antibody. Cancer Res 1991; 51:5425 - 5429
- Denton GW, Durrant LG, Hardcastle JD, Austin EB, Sewell HF, Robins RA. Clinical outcome of colorectal cancer patients treated with human monoclonal anti-idiotypic antibody. Int J Cancer 1994; 57:10 - 14
- Durrant LG, Buckley TJ, Denton GW, Hardcastle JD, Sewell HF, Robins RA. Enhanced cell-mediated tumor killing in patients immunized with human monoclonal antiidiotypic antibody 105AD7. Cancer Res 1994; 54:4837 - 4840
- Durrant LG, Buckley DJ, Robins RA, Spendlove I. 105Ad7 cancer vaccine stimulates anti-tumour helper and cytotoxic T-cell responses in colorectal cancer patients but repeated immunisations are required to maintain these responses. Int J Cancer 2000; 85:87 - 92
- Durrant LG, Maxwell-Armstrong C, Buckley D, Amin S, Robins RA, Carmichael J, Scholefield JH. A neoadjuvant clinical trial in colorectal cancer patients of the human antiidiotypic antibody 105AD7, which mimics CD55. Clin Cancer Res 2000; 6:422 - 430
- Spendlove L, Li L, Potter V, Christiansen D, Loveland BE, Durrant LG. A therapeutic human anti-idiotypic antibody mimics CD55 in three distinct regions. Eur J Immunol 2000; 30:2944 - 2953
- Maxwell-Armstrong CA, Durrant LG, Robins RA, Galvin AM, Scholefield JH, et al. Increased activation of lymphocytes infiltrating primary colorectal cancers following immunisation with the anti-idiotypic monoclonal antibody 105AD7. Gut 1999; 45:593 - 598
- Maxwell-Armstrong CA, Durrant LG, Scholefield JH. Immunotherapy for colorectal cancer. Am J Surg 1999; 177:344 - 348
- Amin S, Robins RA, Maxwell-Armstrong CA, Scholefield JH, Durrant LG. Vaccine-induced apoptosis: a novel clinical trial end point?. Cancer Res 2000; 60:3132 - 3136
- Durrant LG, Parsons T, Moss R, Spendlove I, Carter G, Carr F. Human anti-idiotypic antibodies can be good immunogens as they target FC receptors on antigen-presenting cells allowing efficient stimulation of both helper and cytotoxic T-cell responses. Int J Cancer 2001; 92:414 - 420
- Parsons T, Spendlove I, Nirula R, Writer M, Carter G, Carr F, Durrant LG. A novel CEA vaccine stimulates T cell proliferation, gammaIFN secretion and CEA specific CTL responses. Vaccine 2004; 22:3487 - 3894
- Ullenhag GJ, Spendlove I, Watson NF, Indar AA, Dube M, Robins RA, et al. A neoadjuvant/adjuvant randomized trial of colorectal cancer patients vaccinated with an antiidiotypic antibody, 105AD7, mimicking CD55. Clin Cancer Res 2006; 12:7389 - 7396
- Ullenhag GJ, Spendlove I, Watson NF, Kallmeyer C, Pritchard-Jones K, Durrant LG. T-cell responses in osteosarcoma patients vaccinated with an anti-idiotypic antibody, 105AD7, mimicking CD55. Clin Immunol 2008; 128:148 - 154
- Durrant LG, Denton GW, Jacobs E, Mee M, Moss R, Austin EB, et al. An idiotypic replica of carcinoembryonic antigen inducing cellular and humoral responses directed against human colorectal tumours. Int J Cancer 1992; 50:811 - 816
- Zaghouani H, Steinman R, Nonacs R, Shah H, Gerhard W, Bona C. Presentation of a viral T cell epitope expressed in the CDR3 region of a self immunoglobulin molecule. Science 1993; 259:224 - 247
- Zanetti M, Rossi F, Lanza P, Filaci G, Lee RH, Billetta R. Theoretical and practical aspects of antigenized antibodies. Immunol Rev 1992; 130:125 - 150
- Amzel LM, Poljak RJ. Three-dimensional structure of immunoglobulins. Ann Rev Biochem 1979; 48:961 - 997
- Padlan EA. Anatomy of the antibody molecule. Mol Immunol 1994; 31:169 - 217
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901 - 917
- Kabat EA, Wu TT, Bilofsky H. Unusual distributions of amino acids in complementarity-determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. J Biol Chem 1977; 252:6609 - 6616
- Padlan EA. Structural implications of sequence variability in immunoglobulins. PNAS 1977; 74:2551 - 2555
- Padlan EA, Abergel C, Tipper JP. Identification of specificity-determining residues in antibodies. Faseb J 1995; 9:133 - 139
- Studnicka GM, Soares S, Better M, Williams RE, Nadell R, Horwitz AH. Human-engineered monoclonal antibodies retain full specific binding activity by preserving non-CDR complementarity-modulating residues. Prot Eng 1994; 7:805 - 814
- Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 1997; 273:927 - 948
- Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, et al. Conformations of immunoglobulin hypervariable regions. Nature 1989; 342:877 - 883
- Tramontano A, Chothia C, Lesk AM. Framework residue 71 is a major determinant of the position and conformation of the second hypervariable region in the VH domains of immunoglobulins. J Mol Biol 1990; 215:175 - 182
- Sollazzo M, Billetta R, Zanetti M. Expression of an exogenous peptide epitope genetically engineered in the variable domain of an immunoglobulin: implications for antibody and peptide folding. Prot Eng 1990; 4:215 - 220
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. PNAS 1979; 76:4350 - 4354
- Billetta R, Hollingdale MR, Zanetti M. Immunogenicity of an engineered internal image antibody. PNAS 1991; 88:4713 - 4717
- Brumeanu TD, Swiggard WJ, Steinman RM, Bona CA, Zaghouani H. Efficient loading of identical viral peptide onto class II molecules by antigenized immunoglobulin and influenza virus. J Exp Med 1993; 178:1795 - 1799
- Li S, Polonis V, Isobe H, Zaghouani H, Guinea R, Moran T, Bona C, Palese P. Chimeric influenza virus induces neutralizing antibodies and cytotoxic T cells against human immunodeficiency virus type 1. J Virol 1993; 67:6659 - 6666
- Kuzu Y, Kuzu H, Zaghouani H, Bona C. Priming of cytotoxic T lymphocytes at various stages of ontogeny with transfectoma cells expressing a chimeric Ig heavy chain gene bearing an influenza virus nucleoprotein peptide. Int Immunol 1993; 5:1301 - 1307
- Zaghouani H, Kuzu Y, Kuzu H, Brumeanu TD, Swiggard WJ, Steinman RM, Bona CA. Contrasting efficacy of presentation by major histocompatibility complex class I and class II products when peptides are administered within a common protein carrier, self immunoglobulin. Eur J Immunol 1993; 23:2746 - 2750
- Wallace PK, Tsang KY, Goldstein J, Correale P, Jarry TM, Schlom J, et al. Exogenous antigen targeted to FcgammaRI on myeloid cells is presented in association with MHC class I. J Immunol Methods 2001; 248:183 - 194
- Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. J Exp Med 2002; 195:1653 - 1659
- Ravetch JV. A full complement of receptors in immune complex diseases. J Clinical Investigation 2002; 110:1759 - 1761
- Melief CJ. Mini-review: Regulation of cytotoxic T lymphocyte responses by dendritic cells: peaceful coexistence of cross-priming and direct priming?. Eur J Imunol 2003; 33:2645 - 2654
- Strawbridge AB, Blum JS. Autophagy in MHC class II antigen processing. Curr Opin Immunol 2007; 19:87 - 92
- Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med 1999; 189:371 - 380
- Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest 2005; 115:2914 - 2923
- Fanger NA, Wardwell K, Shen L, Tedder TF, Guyre PM. Type I (CD64) and type II (CD32) Fcgamma receptor-mediated phagocytosis by human blood dendritic cells. J Immunol 1996; 157:541 - 548
- Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, et al. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. PNAS 2005; 102:2910 - 2915
- Bot A, Smith D, Phillips B, Bot S, Bona C, Zaghouani H. Immunologic control of tumors by in vivo Fcgamma receptor-targeted antigen loading in conjunction with double-stranded RNA-mediated immune modulation. J Immunol 2006; 176:1363 - 1374
- Steitz J, Bruck J, Steinbrink K, Enk A, Knop J, Tuting T. Genetic immunization of mice with human tyrosinase-related protein 2: implications for the immunotherapy of melanoma. Int J Cancer 2000; 86:89 - 94
- Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med 2001; 194:823 - 832
- van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 1999; 190:355 - 366
- van Elsas A, Sutmuller RP, Hurwitz AA, et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J Exp Med 2001; 194:481 - 489
- Condon C, Watkins SC, Celluzzi CM, Thompson K, Falo LD Jr. DNA-based immunization by in vivo transfection of dendritic cells. Nat Med 1996; 2:1122 - 1128
- Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J Exp Med 1998; 188:1075 - 1082
- Klinman DM, Sechler JM, Conover J, Gu M, Rosenberg AS. Contribution of cells at the site of DNA vaccination to the generation of antigen-specific immunity and memory. J Immunol 1998; 160:2388 - 2392
- Cho JH, Youn JW, Sung YC. Cross-priming as a predominant mechanism for inducing CD8(+) T cell responses in gene gun DNA immunization. J Immunol 2001; 167:5549 - 5557
- Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules?. J Immunol 1996; 157:1823 - 1826
- Yewdell JW, Nicchitta CV. The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol 2006; 27:368 - 373
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000; 404:770 - 774
- Cawthon AG, Alexander-Miller MA. Optimal colocalization of TCR and CD8 as a novel mechanism for the control of functional avidity. J Immunol 2002; 169:3492 - 3498
- Luxembourg A, Evans CF, Hannaman D. Electroporation-based DNA immunisation: translation to the clinic. Expert Opin Biol Ther 2007; 7:1647 - 1664
- Tjelle TE, Rabussay D, Ottensmeier C, Mathiesen I, Kjeken R. Taking electroporation-based delivery of DNA vaccination into humans: a generic clinical protocol. Methods Mol Biol 2008; 423:497 - 507
- Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003; 421:852 - 856
- Wang JC, Livingstone AM. Cutting edge: CD4+ T cell help can be essential for primary CD8+ T cell responses in vivo. J Immunol 2003; 171:6339 - 6343
- Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, Scott B. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol 2000; 165:6047 - 6055
- Wong SB, Bos R, Sherman LA. Tumor-specific CD4+ T cells render the tumor environment permissive for infiltration by low-avidity CD8+ T cells. J Immunol 2008; 180:3122 - 3131
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157:105 - 132