
Abstract
Alpha-1 antitrypsin (AAT) is a serum protease inhibitor that belongs to the serpin superfamily. Mutations in AAT are associated with α-1 antitrypsin deficiency (AATD), a rare genetic disease with two distinct manifestations: AATD lung disease and AATD liver disease. AATD lung disease is caused by loss-of-function of AAT and can be treated with plasma-derived AAT. AATD liver disease is due to the aggregation and retention of mutant AAT protein in the liver; the only treatment available for AATD liver disease is liver transplantation. Here we demonstrate that antisense oligonucleotides (ASOs) targeting human AAT efficiently reduce levels of both short and long human AAT transcript in vitro and in transgenic mice, providing a novel therapy for AATD liver disease. In addition, ASO-mediated depletion of mouse AAT may offer a useful animal model for the investigation of AATD lung disease.
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a rare hereditary disorder that is characterized by reduced levels of circulating α-1 antitrypsin (AAT). AATD has two distinct manifestations: AATD lung disease and AATD liver disease.Citation1-Citation4 Since its discovery about 50 years ago,Citation5 significant progress has been made in understanding the genetic, molecular, and biochemical characteristics of the disease. AAT is a serum protease inhibitor that belongs to the serpin family. One of the main substrates of AAT is neutrophil elastase (NE). In AATD patients who have reduced AAT levels, NE becomes overactive, leading to the destruction of lung connective tissue and eventually emphysema. Therefore, AATD lung patients are treated by intravenous infusion of plasma-derived AAT.Citation6 AATD lung disease represents a success story in which the discovery of the genetic cause of a rare disease led to development of a treatment.
AAT is predominantly produced by hepatocytes and secreted into blood; but low levels of AAT expression are also detected in the lung and in the gut. AATD liver disease is caused by the aggregation and retention of mutant AAT protein in the liver. Although more than 100 AAT mutations have been identified, Glu342Lys mutation (known as Z mutation) is observed most frequently in the clinic. It is estimated that over 90% of clinical cases are patients homozygous for Z mutation (known as PiZZ patients). The Z mutation changes the conformation of AAT and makes it prone to aggregation. AAT protein aggregates are retained in endoplasmic reticulum (ER), leading to ER stress, liver injury, and eventually fibrosis and hepatocellular carcinoma (HCC) in some PiZZ patients. Currently liver transplantation is the only available treatment for AATD liver disease.Citation2,Citation7-Citation10
Animal models are critical tools in research and in the development of novel therapies, especially in the rare disease arena. In the case of AATD liver disease, multiple transgenic models have been described.Citation11-Citation14 Among these models, PiZ mice contain a human genomic AAT transgene with the Z mutation, driven by its endogenous promoter. PiZ transgene expression mimics its expression in humans, and most importantly, Z protein aggregates are observed in livers of these mice, similar to that observed in PiZZ liver patients. In addition, as PiZ mice also express AAT from endogenous AAT (or protease inhibitor, Pi) genes, these mice do not have lung disease, making these mice a good model for the investigation of AATD liver disease.Citation12 Humans have only one functional AAT gene; mice have three to five AAT genes depending on strain background, possibly due to gene duplication events. Complete deletion of mouse AAT genes leads to embryonic lethality, an indication of the critical roles of AAT in early development in mouse.Citation15,Citation16 The pallid mouse harbors a naturally occurring mutation in the pallid gene, which in turn hinders the normal secretion of AAT and results in decreased circulating AAT levels. These mice display emphysema-like symptoms late in life in C57BL/6 background.Citation17,Citation18 However, circulating AAT levels in this model are reduced by only about 50% compared with those in wild-type mice. A mouse model that allows titration of AAT levels would provide a better understanding of AATD lung disease.
The antisense technology platform enables rapid identification of specific inhibitors of RNA expression of virtually any gene.Citation19 An antisense oligonucleotide (ASO) hybridizes to its complementary RNA target and typically triggers target cleavage by RNase H; degradation of the mRNA prevents production of disease-causing protein. In our recently published work,Citation20 we identified potent ASOs targeting human AAT. Systemic treatment of PiZ mice with AAT-ASO led to significant reductions in circulating levels of AAT and both soluble and aggregated AAT protein in the liver. As a result, AAT-ASO treatment prevents AATD liver disease in young PiZ mice, short-term treatment stops liver disease progression, and long-term treatment reverses disease. Furthermore, ASO treatment markedly decreased liver fibrosis in this mouse model. Administration of AAT-ASO in non-human primates led to reductions in circulating normal AAT of approximately 80%, demonstrating the potential utility of this approach in higher species. Antisense oligonucleotides thus represent a promising therapy for AATD liver disease.
Here we report that alternative polyadenylation of human AAT occurs in humans and in PiZ mice. AAT-ASO reduces both short and long hAAT transcript in vitro and in vivo. We also developed ASOs that reduce levels of the mouse AAT, which may be useful to create a new animal model for the investigation of AATD lung disease.
Human AAT Transcripts are Alternatively Polyadenylated
Analysis of the human AAT gene sequence and expressed sequence tags (ESTs) revealed two potential polyadenylation sites (). Previous studies indicated that a short transcript (~1.4 kb) predominates in HepG2 cells.Citation21 To confirm this finding, ~60 ASOs targeting upstream and downstream of the first polyadenylation sites were synthesized and hAAT mRNA levels in ASO-treated HepG2 cells were measured using a primer probe set (5′ppset) that detects both long and short transcripts. ASOs targeting 5′ of the first poly A site were very active; while those targeting 3′ of the first polyadenylation site showed no activity (). When PCR primers detecting only the long transcript (3′ppset) were used in the analysis, both ASO3 (targeting 5′ of the first polyadenylation site) and ASO4 (targeting 3′ of the first polyadenylation site) resulted in similar reductions. However, when the levels of both long and short transcripts were measured using 5′ppset, ASO3 treatment caused ~80% target reduction, whereas ASO4 had no significant activity (). To further understand this phenomenon, we compared expression levels of hAAT detected by these two primer sets in human liver, HepG2 cells, and Hep3B cells. As shown in , Ct values were consistently lower by 6–8 cycles when both transcripts were detected than when only the long transcript levels were evaluated. These data indicate that most transcripts present in human liver and HCC cell lines are processed at the first polyadenylation site. For subsequent ASO screens, we focused only on ASOs targeting both transcripts and identified potent compounds that display dose-dependent reduction of all AAT transcripts.Citation20
Figure 1. Alternative polyadenylation in human AAT transcripts. (A) Schematic representation of long and short transcripts. The relative locations of ASO3, ASO4, 5′ppset, 3′ppset, and Northern probe are shown (not to scale). (B) Levels of AAT in HepG2 cells were measured with 5′ppset, which detects both long and short transcripts. X axis represents the position of ASOs on the transcript around the first polyadenylation site and Y axis represents AAT mRNA levels after ASO treatment relative to untreated control. (C) Reduction of AAT in HepG2 cells treated with AS03 or AS04 measured with 5′ppset and with 3′ppset relative to untreated control. Cells were electroporated in growth medium in the presence of 20 μM ASO and plated. Twenty-four hours after transfection total cellular RNA was isolated and the amount of AAT mRNA was quantitated using a qRT-PCR assay (TaqMan).
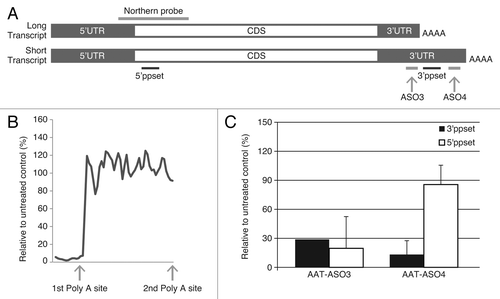
Table 1. Ct values of AAT transcripts detected with 5′ppset and 3′ppset
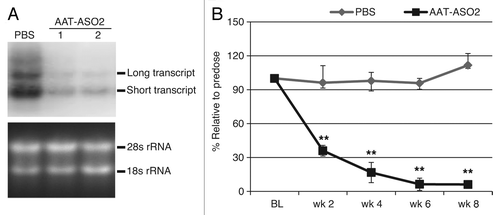
To determine whether alternative polyadenylation of human AAT also exists in PiZ mice and to confirm the effect of ASO treatment, we treated PiZ mice with 50 mg/kg/week AAT-ASO2Citation20 via subcutaneous injection for 8 wk. Liver RNA was extracted and analyzed by northern blot with a probe recognizing both transcripts (). As shown in , both transcripts were detected in PiZ livers, and the short transcript was more abundant than the long transcript. After AAT-ASO2 treatment, levels of both transcripts were reduced significantly. Similar results using the 5′ppset were reported previously.Citation20 Moreover, ASO2 treatment resulted in a dramatic time-dependent protein reduction (), further confirming that both transcripts were almost completely eliminated.
Figure 2. AAT-ASO2 treatment dramatically reduces both long and short transcripts in PiZ mice. (A) northern blot analysis for hAAT in PiZ mice with and without AAT-ASO2 treatment. Northern probe recognizes both transcripts. (B) Time-dependent hAAT reduction in plasma. Six-week-old PiZ mice were treated for 8 wk with 50 mg/kg AAT-ASO2 via subcutaneous injection. Human AAT mRNA levels in liver were quantified by qRT-PCR (TaqMan) using 5′ppset, and plasma AAT levels were determined by an immunoturbidmetric method. Results represent means ± standard deviations (n = 4). **P < 0.01 by two-way ANOVA with Bonferroni’s post-hoc tests.
A Potential Novel AATD Lung Disease Model
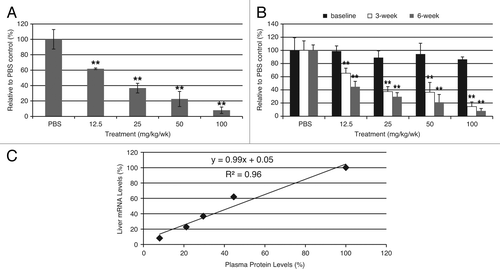
In order to establish a potential mouse model for the investigation of AATD lung disease, we developed ASOs targeting mouse AAT to reduce endogenous AAT levels, as observed in AATD lung patients. Unlike humans who have one AAT or SERPINA1 gene, mice contain 3–5 distinct protease inhibiting genes (SERPINA1A through SERPINA1E, referred to as mAAT as a family in this report), likely due to a gene duplication event.Citation22 mAAT genes share high sequence identity so ASOs targeting all 5 genes were designed and tested using qRT-PCR primers that also match all 5 genes (forward primer: 5′-TTCTGGCAGG CCTGTGTTG -3′; reverse primer: 5′-ATCCTTCTGG GAGGTGTCTG TCT-3′; probe: 5′-CCCCAGCTTT CTGGCTGAGG ATGTTC-3′). Potent ASOs were identified through in vitro and in vivo screens, and one such compound (mAAT-ASO) was evaluated in CD-1 mice. Mice were treated with PBS or with 6.25, 12.5, 25, or 50 mg/kg per week mAAT-ASO for 6 weeks by subcutaneous injection. Dose-dependent reduction in liver mRNA and plasma protein levels were observed with a good correlation (slope = 0.99 and R2 = 0.96, ). With mAAT-ASO treatment, AAT levels can be manipulated as desired; a low dose treatment would result in small reductions while a high dose treatment would dramatically reduce AAT from circulation (e.g., >92% reduction relative to wild-type levels in 100 mg/kg treatment group). These AAT levels could represent the different levels of AAT in AATD patient with various genetic mutations.Citation23
Figure 3. ASO treatment greatly reduced AAT levels in CD-1 mice. (A) Dose-dependent reduction of mouse AAT mRNA levels in CD-1 mice after mAAT-ASO treatment. (B) Dose-dependent reduction of circulating mouse AAT in CD-1 mice after mAAT-ASO treatment. mAAT protein was measured with an ELISA method according to manufacturer’s recommendations (Alpco 41-A1AMS-E01). (C) Correlation between liver AAT mRNA reduction and plasma protein reduction. Six-week-old CD-1 mice were treated for 6 wk with the indicated doses of mAAT-ASO via subcutaneous injection. Mouse AAT mRNA levels in liver were quantified by qRT-PCR (TaqMan) and plasma AAT levels were determined by an ELISA method (Alpco 41-A1AMS-E01). Results represent means ± standard deviations (n = 4). **P < 0.01 by one-way ANOVA with Tukey’s comparisons for panel A and repeated measures two-way ANOVA with Bonferroni’s post-hoc tests for panel B.
Plasma neutrophil elastase activity was evaluated in control and treated mice (using EnzChek Elastase Assay Kit from Molecular Probes). However, the levels of mouse plasma NE levels were below the limit the detection, even in mice with reduced mAAT levels. No gross abnormalities were observed in mice treated with mAAT-ASO. Lung morphology, examined microscopically, remained normal indicating that mAAT reduction in the short-term is well tolerated. Long-term mAAT reduction, possibly in combination with smoking, bleomycin, or in PiZ mice, may likely cause lung phenotypes that resemble AATD lung disease. NE activity can be evaluated before and after lung disease development in the lung where hyperactivity is expected. In addition, a short-term inflammatory stimulus, such as LPS exposure, could be used to evaluate neutroplilic inflammationCitation24 with mATT-ASO treatment.
Discussion
Fifty years after the discovery of AATD by Laurell and Eriksson,Citation5 tremendous progress has been made in the understanding of both emphysema and liver disease.Citation25-Citation27 The lung manifestation of AATD can be treated by intravenous infusion of plasma-derived AAT, although a more convenient treatment is desired. Patients with AATD liver disease still have no treatment options other than liver transplantation. Substantial hurdles exist in the development of novel treatments for AATD liver disease and to improve existing therapies for AATD lung disease. The major hurdles include (1) under diagnosis of AATD, (2) presence of genetic and environmental modifiers that affect disease manifestation in AATD patients, and (3) poor understanding of the natural history especially for AATD liver disease.Citation2,Citation10,Citation27-Citation29 Furthermore, many PiZZ patients live normal lives without any clinical manifestations of AATD. Hence, addressing these key hurdles will provide valuable information to help determine which patients should be treated with AAT-ASO to ameliorate liver disease and how lung disease should be monitored in a clinical trial setting where circulating mutant AAT levels would be further reduced.
The PiZ mutation is believed to have arisen about 2000 years ago in the Viking population.Citation30 PiZZ frequencies in US and in Europe are estimated to be about 1 in 2000 and 1 in 2500, respectively. Despite the multiple biomedical and genetic diagnostic tools available for AATD, it is still significantly under diagnosed. For example, only approximately 10% PiZZ liver patients in the US are diagnosed.Citation10 AATD liver disease affects both PiZZ children and adults. About 10% of PiZZ infants present neonatal jaundice and cholestasis with a subset progressing to advanced fibrosis or cirrhosis that requires liver transplant. Liver disease manifests as slowly progressive fibrosis in adults, associated with increased risk of cirrhosis and HCC. Over one third of adult PiZZ patients have cirrhosis at the time of death; however, most PiZZ patients have fairly normal liver function tests with no or minimal symptoms of liver disease.Citation28 Both genetic and environmental factors are believed to affect the onset, severity and likely progression of the disease, of which we still do not have a clear understanding. Furthermore, natural history information on AATD liver disease is extremely limited.Citation31 In order to define a clear clinical development path for any novel AATD liver disease therapy, including antisense therapy, both identifying the appropriate patient population and understanding the rate of disease progression are essential. In addition, improvement in biomarkers and noninvasive methods to monitor liver disease progression would be instrumental for successful future AATD liver disease clinical trials. Ongoing natural history studies in children (LOGIC, a longitudinal study of genetic causes of intrahepatic cholestasis, NCT00571272) and in adults (http://www.slu.edu/x68167.xml) will hopefully shed light on AATD liver disease prevalence, severity, and progression. These studies may also provide information on both genetic and environmental modifiers of the liver disease.
Augmentation therapy is available to AATD lung patients in many countries including the US, although a more efficacious and convenient therapy would be ideal. Partly due to the lack of an appropriate AATD lung disease model, limited efforts have been dedicated to the development of an improved therapy for AATD lung disease. Using an ASO targeting the endogenous mouse AAT genes, we demonstrated that the steady-state levels of mAAT can be titrated precisely. Reduction in levels of endogenous AAT in the short-term study (6 weeks) did not cause any obvious lung abnormalities. As is observed in AATD patients, chronic mAAT reduction is likely required for lung disease development and smoking or other lung insults will potentially accelerate this process.Citation32 Furthermore, depletion of endogenous AAT in PiZ mice with mAAT-ASO treatment would provide an opportunity to investigate the role of Z protein aggregates in promoting lung disease.Citation33
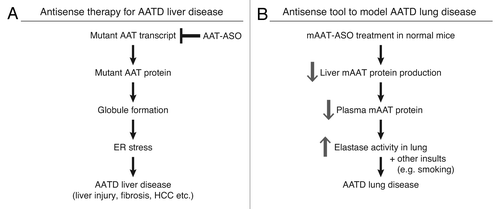
In summary, we have developed potent ASOs for the inhibition of AAT. In PiZ mice, treatment with an ASO that targets the human AAT mRNA significantly ameliorated liver disease.Citation20 The ASO evaluated here that targets the mouse AAT can be used to reduce AAT levels in mice and could enable the creation of an AATD lung disease model (). This work demonstrates that antisense technology can be employed to treat rare disease like AATD liver disease and also used as a research tool for the understanding of disease mechanisms.
Figure 4. Antisense ASOs provide a novel therapy for AATD liver disease and a unique model for AATD lung disease. (A) hAAT-ASO treatment in PiZ mice reduces mutant protein production and aggregation. This decreases ER stress and ameliorates liver disease including cell death, fibrosis, and risk of HCC. (B) mAAT-ASO treatment can be used to model AATD lung disease. mAAT-ASO treatment reduces circulating AAT levels and results in overactive elastase in the lung, which may lead to emphysema in the presence of other insults such as smoking.
Disclosure of Potential Conflicts of Interest
S.G., S.B., A.W., L.A., S.M.F., M.L.M., and B.P.M. are employees and stockholders of Isis Pharmaceuticals. J.H.T has nothing to disclose.
References
- EkeowaUI, GooptuB, BelorgeyD, HägglöfP, Karlsson-LiS, MirandaE, PérezJ, MacLeodI, KrogerH, MarciniakSJ, et al. alpha1-Antitrypsin deficiency, chronic obstructive pulmonary disease and the serpinopathies. Clin Sci (Lond)2009; 116:837 - 50; http://dx.doi.org/10.1042/CS20080484; PMID: 19426146
- FairbanksKD, TavillAS. Liver disease in alpha 1-antitrypsin deficiency: a review. Am J Gastroenterol2008; 103:2136 - 41, quiz 2142; http://dx.doi.org/10.1111/j.1572-0241.2008.01955.x; PMID: 18796107
- FregoneseL, StolkJ. Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Orphanet J Rare Dis2008; 3:16; http://dx.doi.org/10.1186/1750-1172-3-16; PMID: 18565211
- JanciauskieneSM, BalsR, KoczullaR, VogelmeierC, KöhnleinT, WelteT. The discovery of α1-antitrypsin and its role in health and disease. Respir Med2011; 105:1129 - 39; http://dx.doi.org/10.1016/j.rmed.2011.02.002; PMID: 21367592
- LaurellCB, ErikssonS. The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. 1963. COPD2013; 10:Suppl 13 - 8; http://dx.doi.org/10.3109/15412555.2013.771956; PMID: 23527532
- StockleyRA, MiravitllesM, VogelmeierC, Alpha One International Registry (A.I.R.). Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis2013; 8:149; http://dx.doi.org/10.1186/1750-1172-8-149; PMID: 24063809
- ErikssonS, CarlsonJ, VelezR. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N Engl J Med1986; 314:736 - 9; http://dx.doi.org/10.1056/NEJM198603203141202; PMID: 3485248
- LomasDA, EvansDL, FinchJT, CarrellRW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature1992; 357:605 - 7; http://dx.doi.org/10.1038/357605a0; PMID: 1608473
- RudnickDA, PerlmutterDH. Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology2005; 42:514 - 21; http://dx.doi.org/10.1002/hep.20815; PMID: 16044402
- TeckmanJH, LindbladD. Alpha-1-antitrypsin deficiency: diagnosis, pathophysiology, and management. Curr Gastroenterol Rep2006; 8:14 - 20; http://dx.doi.org/10.1007/s11894-006-0059-8; PMID: 16510030
- AliR, PerfumoS, della RoccaC, AmiconeL, PozziL, McCullaghP, Millward-SadlerH, EdwardsY, PoveyS, TripodiM. Evaluation of a transgenic mouse model for alpha-1-antitrypsin (AAT) related liver disease. Ann Hum Genet1994; 58:305 - 20; http://dx.doi.org/10.1111/j.1469-1809.1994.tb00728.x; PMID: 7864587
- CarlsonJA, RogersBB, SifersRN, FinegoldMJ, CliftSM, DeMayoFJ, BullockDW, WooSL. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J Clin Invest1989; 83:1183 - 90; http://dx.doi.org/10.1172/JCI113999; PMID: 2784798
- DycaicoMJ, GrantSG, FeltsK, NicholsWS, GellerSA, HagerJH, PollardAJ, KohlerSW, ShortHP, JirikFR, et al. Neonatal hepatitis induced by alpha 1-antitrypsin: a transgenic mouse model. Science1988; 242:1409 - 12; http://dx.doi.org/10.1126/science.3264419; PMID: 3264419
- RütherU, TripodiM, CorteseR, WagnerEF. The human alpha-1-antitrypsin gene is efficiently expressed from two tissue-specific promotors in transgenic mice. Nucleic Acids Res1987; 15:7519 - 29; http://dx.doi.org/10.1093/nar/15.18.7519; PMID: 3498933
- Kushi A, Akiyama K, Noguchi M, Edamura K, Yoshida T, Sasai H. Disruption of the murine alpha1-antitrypsin/PI2 gene. Experimental animals / Japanese Association for Laboratory Animal Science 2004; 53:437-43.
- WangD, WangW, DawkinsP, PatersonT, KalshekerN, SallenaveJM, HoughtonAM. Deletion of Serpina1a, a murine α1-antitrypsin ortholog, results in embryonic lethality. Exp Lung Res2011; 37:291 - 300; http://dx.doi.org/10.3109/01902148.2011.554599; PMID: 21574874
- MartoranaPA, BrandT, GardiC, van EvenP, de SantiMM, CalzoniP, MarcolongoP, LungarellaG. The pallid mouse. A model of genetic alpha 1-antitrypsin deficiency. Lab Invest1993; 68:233 - 41; PMID: 8441253
- YoshidaM, SakiyamaS, KenzakiK, TobaH, UyamaK, TakehisaM, KondoK, TangokuA. Functional evaluation of pallid mice with genetic emphysema. Lab Invest2009; 89:760 - 8; http://dx.doi.org/10.1038/labinvest.2009.34; PMID: 19381131
- BennettCF, SwayzeEE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol2010; 50:259 - 93; http://dx.doi.org/10.1146/annurev.pharmtox.010909.105654; PMID: 20055705
- GuoS, BootenSL, AghajanM, HungG, ZhaoC, BlomenkampK, GattisD, WattA, FreierSM, TeckmanJH, et al. Antisense oligonucleotide treatment ameliorates alpha-1 antitrypsin-related liver disease in mice. J Clin Invest2014; 124:251 - 61; http://dx.doi.org/10.1172/JCI67968; PMID: 24355919
- PerlmutterDH, ColeFS, KilbridgeP, RossingTH, ColtenHR. Expression of the alpha 1-proteinase inhibitor gene in human monocytes and macrophages. Proc Natl Acad Sci U S A1985; 82:795 - 9; http://dx.doi.org/10.1073/pnas.82.3.795; PMID: 3871944
- HeitC, JacksonBC, McAndrewsM, WrightMW, ThompsonDC, SilvermanGA, NebertDW, VasiliouV. Update of the human and mouse SERPIN gene superfamily. Hum Genomics2013; 7:22; http://dx.doi.org/10.1186/1479-7364-7-22; PMID: 24172014
- StollerJK, AboussouanLS. Alpha1-antitrypsin deficiency. Lancet2005; 365:2225 - 36; http://dx.doi.org/10.1016/S0140-6736(05)66781-5; PMID: 15978931
- BootsAW, GerloffK, BartholoméR, van BerloD, LedermannK, HaenenGR, BastA, van SchootenFJ, AlbrechtC, SchinsRP. Neutrophils augment LPS-mediated pro-inflammatory signaling in human lung epithelial cells. Biochim Biophys Acta2012; 1823:1151 - 62; http://dx.doi.org/10.1016/j.bbamcr.2012.04.012; PMID: 22575681
- GooptuB, DickensJA, LomasDA. The molecular and cellular pathology of alpha-antitrypsin deficiency. Trends Mol Med2013; http://dx.doi.org/10.1016/j.molmed.2013.10.007; PMID: 24374162
- StockleyRA, TurnerAM. alpha-1-Antitrypsin deficiency: clinical variability, assessment, and treatment. Trends Mol Med2013; http://dx.doi.org/10.1016/j.molmed.2013.11.006; PMID: 24380646
- TeckmanJH, JainA. Advances in alpha-1-antitrypsin deficiency liver disease. Curr Gastroenterol Rep2014; 16:367; http://dx.doi.org/10.1007/s11894-013-0367-8; PMID: 24338605
- ClarkVC, DhanasekaranR, BrantlyM, RouhaniF, SchreckP, NelsonDR. Liver test results do not identify liver disease in adults with α(1)-antitrypsin deficiency. Clin Gastroenterol Hepatol2012; 10:1278 - 83; http://dx.doi.org/10.1016/j.cgh.2012.07.007; PMID: 22835581
- TeckmanJH. Liver disease in alpha-1 antitrypsin deficiency: current understanding and future therapy. COPD2013; 10:Suppl 135 - 43; http://dx.doi.org/10.3109/15412555.2013.765839; PMID: 23527737
- LomasDA. The selective advantage of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med2006; 173:1072 - 7; http://dx.doi.org/10.1164/rccm.200511-1797PP; PMID: 16439713
- SvegerT. The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr Scand1988; 77:847 - 51; http://dx.doi.org/10.1111/j.1651-2227.1988.tb10767.x; PMID: 2905108
- ClancyJ, TurnerC. Smoking and COPD: the impact of nature-nurture interactions. Br J Nurs2013; 22:820 - , 822-6; PMID: 24260992
- DickensJA, LomasDA. Why has it been so difficult to prove the efficacy of alpha-1-antitrypsin replacement therapy? Insights from the study of disease pathogenesis. Drug Des Devel Ther2011; 5:391 - 405; PMID: 21966212