Abstract
DEAD-box proteins are superfamily 2 helicases that function in all aspects of RNA metabolism. They employ ATP binding and hydrolysis to generate tight, yet regulated RNA binding, which is used to unwind short RNA helices non-processively and promote structural transitions of RNA and RNA-protein substrates. In the last few years, substantial progress has been made toward a detailed, quantitative understanding of the structural and biochemical properties of DEAD-box proteins. Concurrently, progress has been made toward a physical understanding of the RNA rearrangements and folding steps that are accelerated by DEAD-box proteins in model systems. Here, we review the recent progress on both of these fronts, focusing on the mitochondrial DEAD-box proteins Mss116 and CYT-19 and their mechanisms in promoting the splicing of group I and group II introns.
Introduction
DEAD-box proteins make up the largest group of nucleic acid helicases.Citation1 They comprise a family within helicase superfamily 2 and function in essentially all facets of cellular RNA metabolism, including transcription, mRNA and tRNA processing, protein synthesis, RNA nuclear export and RNA degradation.Citation2,Citation3 Many of these processes involve RNAs with defined structures,Citation4-Citation8 and the most common role of DEAD-box proteins is to use cycles of ATP binding and hydrolysis to promote conformational rearrangements of these RNAs during their biogenesis or in the course of their cellular functions.Citation9,Citation10 Some DEAD-box proteins are directed to individual RNA targets, whereas others function more broadly as general chaperones in RNA folding and RNA-protein assembly processes,Citation11 in line with an early suggestion that the fundamental properties of RNA are likely to lead to a general requirement for chaperones in RNA folding.Citation12
Along with the advances in understanding the physiological roles of DEAD-box proteins, much has been learned about their biochemical properties. Their most fundamental property is that they possess ATPase activity that is stimulated by RNA.Citation13 Another feature that appears to be universal is that the ATPase cycle is linked to a cycle of changes in affinity for RNA.Citation14 These basic properties are used in different ways by different DEAD-box proteins for a broad range of biochemical activities and physiological roles. DEAD-box proteins commonly have the ability to use ATP to unwind short RNA duplexes, as well as additional activities such as disruption or prevention of RNA-protein interactionsCitation15-Citation17 and acceleration of RNA helix formation (RNA strand annealing).Citation18-Citation21
Determining which properties are general for all DEAD-box proteins and which are specific to individual ones is critical for developing a molecular understanding of the functions of these remarkable enzymes. Toward this objective, here we first outline our current understanding of the biochemical properties and activities of DEAD-box proteins, focusing on recent developments in this dynamic field. We then describe how these activities are understood to underlie the ability of DEAD-box proteins to promote RNA structural rearrangements, with an emphasis on the yeast mitochondrial (mt) protein Mss116 and its Neurospora crassa ortholog CYT-19. These proteins function as general RNA chaperones for the folding of mt group I and group II introns and participate in translational activation and other RNA processing reactions.Citation22-Citation24 They have been the focus of intense biochemical and biophysical dissection in recent years and represent well-defined systems for exploring the relationship between the biochemical properties and physiological roles of DEAD-box proteins. We include results delineating biochemical properties of other DEAD-box proteins both to emphasize points of generality and to highlight important differences between different DEAD-box proteins.
Structural characteristics and biochemical activities of DEAD-box proteins
Crystallographic analyses have revealed structural features that are universal to DEAD-box proteins.Citation25-Citation35 Like other superfamily 1 and 2 helicases, they possess a conserved core consisting of two RecA-like domains that are connected by a flexible linker. At least 13 conserved motifs lie within these domains and contribute to interactions between the two domains or with substrates (). Crystal structures of ternary complexes of DEAD-box proteins with a bound adenosine nucleotide analog and small ssRNA have shown that ATP binds to a site that is largely contained within domain 1 but also includes amino acid residues from domain 2. Within domain 1, the triphosphate moiety interacts with motif I, which includes a sequence known as the P-loop or Walker A motif, and motif II, which includes the eponymous D-E-A-D sequence. In addition, the base-pairing face of the adenine nucleotide forms specific interactions with a glutamine of the Q-motif, resulting in a strong preference in DEAD-box proteins for ATP over other nucleoside triphosphates.Citation36,Citation37 Contacts with domain 2 include interactions of the triphosphate with motif VI and the ribose sugar with motif Va. Motif III lies near the γ-phosphoryl group of ATP at the interface of the two domains. On the opposite side of the protein, single-stranded RNA (ssRNA) also binds across the domain interface to a site that includes contacts with motifs Ia, Ib and Ic from domain 1, along with motifs IV, IVa, V and Vb from domain 2.
Figure 1. Core domain structure of DEAD-box proteins. The top panel shows a stereoview of Mss116 bound to AMP-PNP and ssRNA (U10).Citation31 Motifs that interact with ATP are orange and motifs that interact with ssRNA are green. Motif III, which does not contact ATP or ssRNA but is involved with communication between the two domains and with substrates, is olive. DI is dark gray, DII is silver, the linker between them is dark blue and the CTE, present in Mss116 and CYT-19, is light blue. Below the structure is a cylinder representation showing the domains and motifs in the same colors. Also shown are the N-terminal extension (NTE) and the C-tail of Mss116, which were not present or not resolved in the crystal structures (refs. Citation31 and Citation41).
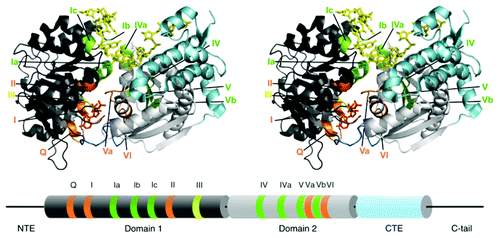
A universal feature of DEAD-box proteins is that bound ssRNA undergoes a sharp bend that is induced by ionic and H-bond interactions with a wedge helix containing motif Ic in domain 1.Citation28-Citation35 The requirement for ssRNA to bind in this conformation, which is incompatible with helix formation, contributes to generating the local strand separation that is used for RNA unwinding.Citation38-Citation40 Additionally, the structure of Mss116 revealed a second bend in the bound ssRNA, resulting in RNA crimping.Citation31 This second bend is caused primarily by a steric block between nucleotides near the 5′ end of the bound ssRNA and a C-terminal extension (CTE) of domain 2.Citation41
In addition to the conserved core, most DEAD-box proteins have extensions or ancillary domains, which differ for different DEAD-box proteins and can play a variety of roles. The ancillary domains of a number of DEAD-box proteins interact with RNA or protein components of substrates, thereby targeting individual DEAD-box proteins to their physiological targets.Citation9,Citation10 An archetypal protein of this type is the bacterial DbpA/YxiN protein, which has an ancillary C-terminal domain that binds specifically to a helix and adjacent structure within the large subunit rRNA.Citation42-Citation44 Ancillary domains can also mediate protein dimerizationCitation45 or regulate the functional properties of the core,Citation34 and in some related proteins they contribute additional enzymatic activities.Citation46,Citation47 In Mss116 and CYT-19, domain 2 contains an α-helical CTE of ~100-amino-acids followed by a positively charged, 60–75 amino acid segment termed the C-tail ().Citation31,Citation48 The CTE stabilizes domain 2 and induces the second RNA bend noted above, resulting in RNA crimping,Citation31 while the C-tail contributes to the non-specific binding of RNA substrates (see below).Citation48,Citation49
Cooperative binding of RNA and adenosine nucleotide
At the heart of the ATP-dependent activities of DEAD-box proteins is a cycle of RNA affinity changes that is tightly linked to the ATPase cycle. The effect of these affinity changes is to generate strong, yet regulated binding to a strand of RNA, displacing this segment from a partner strand and temporarily preventing it from forming intramolecular or intermolecular contacts with other RNA segments or proteins components.
Indeed, many DEAD-box proteins have been shown to bind cooperatively to ssRNA and adenosine nucleotides. Cooperativity was demonstrated for the mammalian eIF4A protein by steady-state ATPase activityCitation14 and for E. coli DbpA and other DEAD-box proteins by binding assays using nucleotide analogs such as AMP-PNP.Citation37,Citation50-Citation53 A critical feature of the ATPase cycle is that the level of cooperativity changes dramatically at different stages, regulating the affinity for RNA. In general, positive cooperativity is associated with the presence of the γ-phosphoryl group of the adenosine nucleotide (). Thus, in the presence of ATP or an ATP analog, binding of RNA is typically tightened. However, a limitation of ATP analogs is that they do not mimic all of the binding properties of ATP, as most do not stimulate RNA unwinding,Citation39,Citation40,Citation54 so it is unclear whether the level of cooperativity is the same as with ATP. In contrast, ADP binding is typically observed to give energetically independent binding of RNACitation55 or anti-cooperativity.Citation14,Citation37
Figure 2. RNA affinity changes resulting from changes in cooperativity during the ATPase cycle. In the absence of nucleotide (No nt), cooperativity is absent by definition. Cooperativity is typically observed in the ATP-bound state and is maximal in the ADP-Pi state, as shown by the thickening blue stripe. Anti-cooperativity in the ADP-bound state is shown by the gray stripe. Specific values of cooperativity for Mss116 are indicated for each state.Citation56 The colors for the nucleotide states match those in .
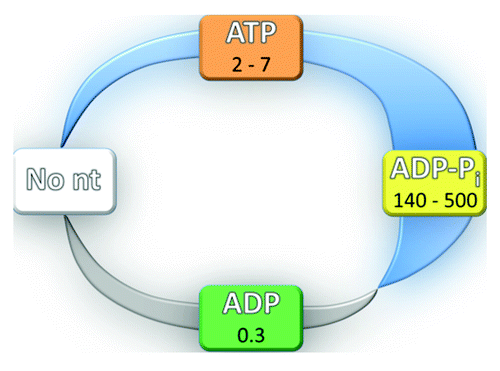
To understand and predict the functional properties of a given DEAD-box protein, it is necessary to have a detailed, quantitative understanding of the energetic coupling between nucleotide and RNA binding at each stage of the ATPase cycle. A key step was taken recently for Mss116 ().Citation56 Fluorescence approaches with a truncated protein construct that includes the tandem RecA-like core domains and the CTE but lacks the C-tail and a short N-terminal extension (NTE) showed that ATP binding increases the affinity for a small double-stranded, hairpin RNA (2–7-fold). In contrast, RNA binding is negatively coupled with ADP binding (3-fold). Further experiments showed that RNA accelerates hydrolysis of ATP and suppresses its re-synthesis on the enzyme, such that there is a dramatic shift of the equilibrium toward products. This knowledge allows calculation of the affinity for RNA when the protein is bound to ADP and Pi (denoted the ADP-Pi state), which indicates very tight RNA binding (140–500-fold tighter than with ATP). The RNA affinity cycle that emerges has important parallels with that derived from the same approaches for DbpA.Citation55 For both proteins, the tightest binding of RNA occurs in the ADP-Pi state and is substantially weaker in the ATP and ADP-bound states. Unlike Mss116, cooperativity with ATP and anti-cooperativity with ADP were not detected for DbpA.
Key to understanding how DEAD-box proteins manipulate RNA structures is a molecular understanding of the conformational changes that occur during the ATPase cycle and underlie the cooperativity between RNA and adenosine nucleotide. All current models link positive cooperativity to the closing of the two core domains.Citation9,Citation10,Citation29 In the absence of substrates, DEAD-box proteins populate conformational ensembles in which the two core domains are separated and have some independent mobility. This property was noted in the earliest crystal structures,Citation25,Citation26 and more recently has been observed for DbpA in fluorescence resonance energy transfer (FRET) experimentsCitation53 and for Mss116 and CYT-19 by small angle X-ray scattering (SAXS).Citation57 Upon binding RNA and ATP or an ATP analog, the domains pack against each other in a ‘closed’ conformation.Citation28-Citation31,Citation33-Citation35 In this closed conformation, both ligands form contacts with both domains and thus the domain closure is highly likely to be the main source of cooperativity.Citation29 The increase in cooperativity in the transition from the ATP to the ADP-Pi state most likely reflects rearrangements that occur upon ATP hydrolysis during or after core closure and affect the affinity for the bound RNA.
There are two key predictions from this model. First, ligand combinations that do not give cooperative binding should not give stable formation of the correct closed conformation. Consistent with this prediction, ADP and RNA binding to DbpA do not give a stable, closed conformation as viewed by FRET.Citation53 Second, the cooperativity of binding might be particularly sensitive to the domain interface. Indeed, mutations in motifs III and Va, both of which line the domain interface, have been observed to reduce the cooperativity without substantially weakening binding of the individual ligands.Citation58,Citation59
ATPase cycle kinetics
Knowledge of the relative and absolute rate constants for individual steps in the ATPase cycle is another key toward understanding how DEAD-box proteins carry out RNA remodeling reactions. This knowledge is necessary to understand which functional states of the DEAD-box protein will accumulate and to relate the time scales necessary for the DEAD-box protein actions to the subsequent RNA folding steps and rearrangements. While the absolute rate constants will differ for different proteins and may depend on the specific RNA substrate and on solution conditions, there are likely to be general themes for the relative rate constants that are useful as a starting point.
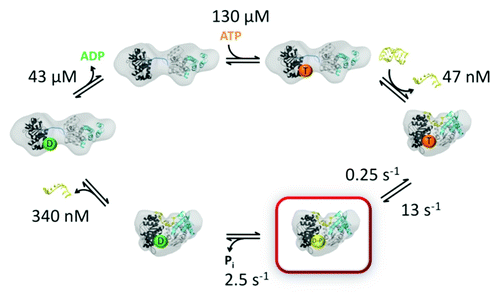
A general picture of the ATPase kinetics of DEAD-box proteins has emerged from extensive data sets for DbpA and Mss116 (),Citation55,Citation56 along with previous work on eIF4A and the yeast protein Ded1, which functions in the nucleus and cytosol in processes including RNA splicing and translation.Citation60 ATP and RNA can bind in either order.Citation14 The affinity for ATP is typically between 50 and 500 µM in the absence of RNACitation14,Citation23,Citation55,Citation56,Citation61 and can vary across this range for different proteins and even for the same protein under different solution conditions.Citation14 In the absence of RNA, ATP hydrolysis is slow (< 0.3 sec–1), such that the equilibrium constant for binding is expected to be equal to the KM measured by steady-state ATPase activity, and thus the affinity estimates above include KM values. Upon binding RNA, ATP hydrolysis is stimulated to 5–10 sec–1, generating an ADP-Pi state that remains bound to RNA. As noted above, RNA affinity is maximized in this state. With bound RNA, ATP hydrolysis is largely irreversible because Pi release is faster than ATP re-synthesis, but release of Pi is slow enough to be at least partially rate-limiting for the overall ATPase cycle (2–6 sec–1). Therefore, under conditions of saturating ATP and RNA, the protein spends a significant fraction of the time in the tight-binding, ADP-Pi state.Citation55,Citation56 Upon release of Pi, the affinities for RNA and ADP are dramatically reduced. ADP is released rapidly (30–100 sec–1), and RNA presumably is as well. At this point, the protein can re-bind weakly to RNA and can again bind ATP to begin another cycle. It may also re-bind ADP, which for some DEAD-box proteins binds substantially tighter than ATP.Citation14,Citation55 In vivo, at cellular concentrations of ADP, the net exchange of ADP for ATP may be partially rate-limiting for the overall cycle for some DEAD-box proteins.Citation55
Figure 3. ATPase kinetics of DEAD box proteins. Specific rate and equilibrium constants are shown for Mss116 (data from ref. Citation56) The domain structures shown are based on SAXS studies of the open and closed complexes.Citation57 Nucleotides are abbreviated by T (ATP), D-Pi (ADP and Pi) and D (ADP). The ADP-Pi state is highlighted in red to indicate that it is populated to the greatest extent in the steady-state with saturating ATP and RNA.
For many DEAD-box proteins, another level of regulation in the ATPase kinetics is conferred by interactions with partner proteins. There is no evidence for such regulation for Mss116 or CYT-19, which can function by themselves as general RNA chaperones in vitro (see RNA remodeling functions of DEAD-box proteins below). On the other hand, a growing number of DEAD-box proteins are known to interact with proteins that modulate the rates of individual steps in the ATPase cycle and thereby influence the timing of assembly and disassembly with RNA. The eIF4A protein interacts with other components of the translation initiation machinery, including eIF4G and eIF4B, and these interactions accelerate the ATPase cycle.Citation62-Citation66 Co-crystal structures and single molecule FRET experiments indicate that the binding of eIF4G promotes a conformation of the two core domains of eIF4A that is neither fully open nor fully closed,Citation64,Citation67 presumably increasing the rates of nucleotide exchange and Pi release.Citation64 Interestingly, Ded1 also forms a complex with eIF4G, other proteins of the eIF4F complex, and mRNA, but the complex is arrested rather than accelerated through the ATPase cycle.Citation68
For two other DEAD-box proteins, recent evidence indicates that interactions with partner proteins tightly regulate the linked RNA-binding and ATPase cycles and are essential for the biological roles of these proteins. The first is eIF4A-III, which functions as a clamp in the exon junction complex (EJC).Citation28,Citation30,Citation33,Citation69 When bound to mRNA and the other protein components of the EJC, Magoh and Y14, eIF4A-III is arrested in the ADP-Pi state, resulting in a very stable complex.Citation33 The protein PYM promotes dissociation of this complex,Citation70 but its effects on the ATPase kinetics of eIF4A-III have not been explored.
Partner proteins are also critical for the ATPase cycle of Dbp5 (DDX19 in humans), which functions at the cytoplasmic face of the nuclear pore to promote export of RNA.Citation17,Citation71-Citation73 Dbp5 interacts strongly with the nucleoporin Nup159 (NUP214 in humans). In addition to localizing Dbp5 at the nuclear pore, this interaction accelerates ADP release by Dbp5, which is suggested to be inherently slow.Citation74 The Gle1 protein, which is also localized at the cytoplasmic face of the nuclear pore, interacts strongly with Dbp5 in the presence of the cofactor IP6. Interestingly, Gle1-IP6 binds to the helicase core at the same site that eIF4G binds to eIF4A, and in the complex the two core domains of Dbp5 are separated and the RNA binding site is disrupted.Citation75 Thus, Gle1-IP6 binding is strongly anti-cooperative with RNA binding, which may result in rapid release of Gle1-IP6 upon binding of RNACitation74 and/or acceleration of RNA release to allow recycling of Dbp5.Citation75 On the other hand, Gle1-IP6 binding is cooperative with nucleotide binding, perhaps contributing to the requirement for Nup159 to accelerate ADP release.Citation74
ATP utilization during RNA unwinding
A key goal for a mechanistic understanding of DEAD-box proteins is to understand how the ATPase cycle is coupled to local RNA unwinding. Although our understanding remains incomplete, considerable progress has been made in the last few years. It was shown for Ded1, Mss116, eIF4A and DbpA/YxiN that complete unwinding of relatively short duplexes can be achieved in the presence of the analog ADP-BeFx.Citation40,Citation54 This finding indicates that unwinding occurs without ATP hydrolysis and that ATP hydrolysis is required primarily to generate the weaker binding states that allow release of the bound RNA strand and recycling of the enzymes.
In a separate study, ATP hydrolysis and RNA unwinding were measured in parallel to determine the number of ATPs consumed during unwinding.Citation39 The implication from the finding above is that unwinding would require a single cycle of ATP binding and hydrolysis, and indeed a stoichiometry of one ATP per duplex unwound was observed for short duplexes under a range of conditions. Together, these results provide strong evidence for a simple coupling between the ATPase cycle and the RNA binding cycle and a mechanism of unwinding that does not involve multiple cycles with directional translocation.Citation38,Citation76 This model is consistent with the known ability of most DEAD-box proteins to unwind short RNA helices but not helices longer than 10–15 bp.Citation18,Citation77,Citation78 It was also shown that more ATP is hydrolyzed during unwinding of longer or more stable helices, indicating that ATP hydrolysis does not always lead to complete unwinding of such duplexes and implying that some ATPase cycles are futile.Citation79 Results suggesting hydrolysis of a single ATP during unwinding of short RNA helices have since been obtained for other DEAD-box proteins and one protein from another SF2 family,Citation56,Citation80,Citation81 suggesting that this general mechanism applies to a range of helicases.
On a molecular level, the model that emerges is that binding of ATP and dsRNA can lead to a conformational change that generates local strand separation. For a short, relatively unstable duplex, this conformational change and resulting strand separation can be sufficient to generate complete unwinding, and the strand that is not tightly bound by the DEAD-box protein dissociates. The conformational change and strand separation would follow ATP and RNA binding and be slower than these processes, but it would be faster than ATP hydrolysis. The rate and extent of this conformational change could also depend on the stability of the RNA duplex, because this would influence the energy barrier that must be overcome. As expected for nucleotide binding followed by a conformational change, multi-phase kinetics have indeed been observed kinetics experiements monitoring nucleotide binding.Citation55,Citation56,Citation64 A recently published paper has provided detailed insight into the structural basis for RNA duplex recognition and unwinding by Mss116 and a comprehensive structural model for RNA unwinding by DEAD-box proteins.Citation82
RNA interactions formed by regions outside the helicase core
In addition to interacting with dsRNA and ssRNA at the canonical RNA binding site within the core, some DEAD-box proteins use additional RNA interactions to localize the core to the physiological targets. Molecular features have been elucidated for two types of interactions. Mss116 and CYT-19 possess at the extreme C-terminus a stretch of 60–75 amino acids termed the C-tail (). This sequence is hydrophilic, rich in arginine and predicted to be unstructured in solution.Citation48 The C-tails of both proteins are easily removed by limited proteolysis, suggesting that they are connected by a relatively accessible linker.Citation49,Citation56 RNA unwinding by CYT-19 is enhanced by RNA structure adjacent to the helix substrate, regardless of whether the extension is ssRNA, dsRNA, dsDNA, or a higher-order structured RNA,Citation83 and this enhancement is dependent on the C-tail.Citation49 These results suggest that the C-tail contacts the adjacent RNA and tethers the core for RNA unwinding.Citation49,Citation83
A recent study using SAXS supports and extends this model.Citation57 For both CYT-19 and Mss116, either alone in solution or with the core bound to ADP-BeFx and a short ssRNA, the scattering profiles indicate that the C-tails occupy space adjacent to domain 2 and are largely unstructured. SAXS data from complexes with nucleic acid constructs that include extensions to the ssRNA showed that the C-tail is flexible and can move through a wide arc to interact with extensions at either end of the ssRNA (). These results suggest that the C-tail provides a flexible tether, which binds non-specifically to large RNA substrates and allows the helicase core to act repeatedly on neighboring regions ().
Figure 4. RNA interactions with regions of DEAD-box proteins outside the helicase core. (A) The C-tails of Mss116 and CYT-19 are unstructured in solution and are flexible, as revealed by SAXS experiments,Citation57 and are able to move relative to the core across a wide region of space. (B) Interactions of Mss116 and CYT-19 with structured RNA. The high flexibility of the C-tail is suggested to allow it to contact structured RNA and to tether the helicase core in proximity to structured RNA, while allowing the core to move to sample different regions of the RNA. The figure shows a model of the Tetrahymena group I intronCitation150 and a model from the solution and crystal structures of Mss116.Citation57 (C) A tethering interaction is formed by the C-terminal domain of DbpA/YxiN with a specific hairpin structure of a 23S rRNA precursor. Panels A and B are reprinted from ref. Citation57 and panel C is reprinted from ref. Citation44 (Copyright 2010), with permission from Elsevier.
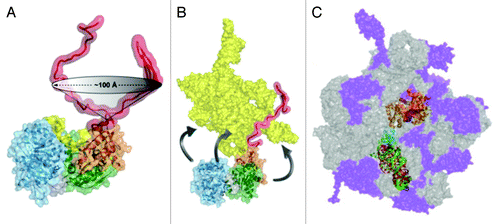
A detailed understanding of the RNA targeting interactions has been achieved for E. coli DbpA and its B. subtilis ortholog YxiN. A C-terminal domain of this protein adopts the fold of an RNA recognition motif (RRM) and binds specifically to a hairpin structure within the 23S rRNA (helix 92), localizing DbpA for participation in biogenesis of the ribosomal large subunit.Citation42,Citation84,Citation85 A co-crystal of the RRM domain of YxiN and an RNA that included helix 92 and two additional helices showed that the RRM domain binds helix 92 and an adjacent junction in a mode different from that observed previously for other RRMs.Citation44 The flexible linkage between the RRM domain and the helicase coreCitation86 may permit the RRM domain to form a tethering interaction while the core interacts with nearby RNA structures, analogous to the tethering interaction of the C-tails of Mss116 and CYT-19 but in this case at a specific location within the 23S rRNA ().
RNA remodeling functions of DEAD-box proteins
DEAD-box proteins facilitate rearrangements of most of the known structured RNAs. These RNAs range from the relatively simple self-splicing introns to complex RNA-protein machines such as the ribosome and spliceosome. Three DEAD-box proteins, along with five proteins from other SF2 families, are required for the extensive rearrangements that occur during the formation and function of the spliceosome.Citation87,Citation88 Analogously, several DEAD-box and related proteins are required for proper folding and assembly of the ribosome. There are excellent recent reviews on the roles of DEAD-box proteins in these complex machines,Citation6,Citation89,Citation90 and here we focus on simpler RNA systems, specifically group I and group II introns. These RNAs have been studied for decades as models of RNA tertiary folding and catalysis, and they allow a broad range of approaches for probing the mechanisms of DEAD-box proteins in facilitating RNA folding processes. A key goal is to develop a molecular understanding that directly and quantitatively connects the biochemical properties of DEAD-box proteins to their physiological roles in these processes. Although we have not yet achieved this goal, work in the last few years has increased our understanding of RNA folding processes that are accelerated by DEAD-box proteins and the biochemical activities that are required in vitro and in vivo.
Group I introns
Group I introns have been valuable model systems for studying the mechanisms of DEAD-box proteins as general RNA chaperones.Citation11,Citation91 These introns are autocatalytic RNAs that consist of a conserved catalytic core and various peripheral structural elements.Citation92,Citation93 The catalytic core consists of two extended helical domains (the P4-P6 and P3-P9 domains),Citation93,Citation94 and these domains interact to form the RNA's active site, which catalyzes a two-step splicing reaction.Citation95,Citation96 The core is stabilized by interactions with and between the peripheral elements, and most group I introns are also stabilized by protein splicing factors that bind specifically to the intron RNA and are required for efficient splicing in vivo.Citation97
In addition to splicing factors, the efficient splicing of several mt group I introns in N. crassa and all group I introns in S. cerevisiae requires the DEAD-box proteins CYT-19 and Mss116, respectively.Citation22-Citation24 In N. crassa, CYT-19 functions in group I intron splicing with the splicing factor CYT-18 (the mt tyrosyl-tRNA synthetase),Citation23 whereas in S. cerevisiae, Mss116 functions in concert with a number of different splicing factors that act on different group I introns.Citation24 In contrast to splicing factors, which bind specifically to the intron RNAs and stabilize the active RNA structure, CYT-19 and Mss116 bind group I intron and other RNAs non-specifically and function primarily to resolve stable inactive or intermediate structures (“kinetic traps”) that limit the rate of RNA folding.Citation11 As expected from their non-specific RNA binding, CYT-19 and Mss116 can readily substitute for each other or be replaced by other DEAD-box proteins, such as Ded1, in group I intron splicing reactions in vitro or in vivo.Citation23,Citation24,Citation98 Studies using a protein-dependent in vitro splicing system for the yeast mt group I intron aI5β showed that Mss116 acting after binding of a structure-stabilizing splicing factor accelerates the second splicing step, exon ligation, for that intron.Citation99
It has been appreciated for decades that RNAs are prone to misfolding, starting from pioneering studies with tRNA and 5S rRNA.Citation100,Citation101 More recently, misfolding has been demonstrated for several group I introns (described further below),Citation102-Citation107 the bacterial RNase P RNA,Citation108 and the hepatitis delta virus ribozyme,Citation109 leading to recognition of misfolding as a dominant theme in RNA folding.Citation91,Citation110 A notable exception was thought to be the Azoarcus group I intron ribozyme, which is roughly half the length of the Tetrahymena ribozyme and forms tertiary structure within milliseconds.Citation111 However, recent studies of that intron revealed one or more intermediates that persist on longer time scalesCitation112 and refold slowly to the native state at 25°C.Citation113
The group I intron from the large subunit rRNA of Tetrahymena thermophila, first shown to be catalytic by Cech and colleagues,Citation114 has been used extensively in studies of RNA folding. Upon addition of Mg2+ in vitro, it was shown to adopt misfolded conformations that included alternative base pairs within the 5′ exon,Citation103,Citation115 and subsequent work provided evidence for non-native structures within the intron as well.Citation116 The RNA chaperone activity of DEAD-box proteins was first demonstrated by showing that CYT-19 could accelerate splicing by resolving kinetic traps in variants of the Tetrahymena intron, and later work showed that this general chaperone function could be performed equally well by Ded1.Citation23,Citation98
To probe folding of the intron portion in greater detail, further studies have used “ribozyme” constructs, in which the exons are removed, leaving an RNA that acts as a true enzyme by binding and cleaving a substrate oligonucleotide in a reaction that mimics the first step of splicing.Citation117 In addition to simplifying the folding process by eliminating effects of exons, such constructs offer the advantage that the fraction of native ribozyme can be determined in a robust and versatile way by measuring the fraction of substrate that is cleaved rapidly upon addition at various folding times.Citation118,Citation119 Chemical footprinting and catalytic activity approaches showed that the standard ribozyme version of the Tetrahymena intron folds through a series of intermediates,Citation120,Citation121 and most of the population folds to a long-lived misfolded intermediate.Citation105,Citation122-Citation125 This misfolded intermediate is extensively structured, including a large amount of native structure and refolds to the native state slowly, on the time scale of hours under typical in vitro conditions.Citation122,Citation126 The refolding requires extensive transient unfolding, which is thought to be necessary to allow a topological change within the ribozyme core.Citation126
Because the misfolded conformation of the Tetrahymena ribozyme is formed by a large fraction of the RNA and is sufficiently long-lived that a nearly homogeneous population can be generated experimentally, its refolding reaction can be readily studied in greater depth. Building on earlier work,Citation23 catalytic activity measurements showed that CYT-19 accelerates refolding of the misfolded ribozyme to the native state and is not needed for subsequent catalytic activity.Citation83 Because the refolding reaction is rate-limited by the transient disruption of tertiary structure,Citation126 CYT-19 is inferred to accelerate the loss of tertiary structure. Consistent with this interpretation, the acceleration depends on ATP and is enhanced by mutations in the ribozyme that weaken tertiary structure.Citation83,Citation127
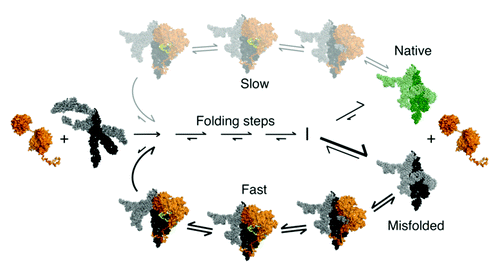
Further work showed that CYT-19 does not preferentially recognize specific structural features of the misfolded RNA and can also unfold the native ribozyme, but the unfolding activity is much less efficient for the native state because this structure is more stable.Citation127 These results suggest a general model for chaperone activity of DEAD-box proteins in which cycles of non-specific RNA unfolding and refolding can give additional chances for folding to the native state, which accumulates if it is more stable than the misfolded state ().
Figure 5. Model for general chaperone activity of DEAD-box proteins. The model is based on refolding of the Tetrahymena group I intron ribozyme by CYT-19. ATP-dependent unfolding of misfolded conformers allows the RNA additional chances to fold to the native state (green). Although the unfolding process mediated by CYT-19 is inherently non-specific, the native state of the RNA can be unfolded with much lower efficiency than misfolded states if it is more stable. The native and misfolded structures of the Tetrahymena ribozyme are shown as identical to emphasize the extensive native structure in the misfolded form of this RNA and because the specific structural difference is not known. A plausible model for the unfolding process is shown, in which CYT-19 first binds to the ribozyme non-specifically by interacting with the C-tail, and then the helicase core engages and unwinds a duplex in an ATP-dependent reaction, which also requires or results in disruption of a tertiary contact. The sequestration and local unwinding of this helix then fosters additional losses of structure, which may propagate along the unwound helix or through space, as shown, by the loss of tertiary structure. Although this model is consistent with the known properties of CYT-19 and the refolding process of this RNA, the specific pathways and mechanisms of unfolding are not known.
Unfolding of the native Tetrahymena ribozyme has also been used as a quantitative assay to probe the mechanisms of RNA unfolding by DEAD-box proteins. It was shown that Mss116, like CYT-19, can unfold the native ribozyme.Citation128 A mutation of motif III of Mss116 (SAT→AAA), which weakens ATP-dependent RNA unwinding by Mss116, also decreases the efficiency of this unfolding reaction and by a similar amount.Citation128 For the wild-type protein, the rate constant for unfolding the ribozyme was at least 0.1 sec–1 and did not reach saturation under the available conditions. Interestingly, this lower limit is within 20-fold of the maximal flux through the ATPase cycle (see ATPase cycle kinetics above).Citation56 This connection to the ATPase measurements provides an important mechanistic insight because it most simply suggests that unfolding of the ribozyme requires no more than 20 ATPase cycles. These ATPase cycles are presumably used to disrupt RNA structural elements, allowing formation of new contacts that lead to formation of the native state. It remains to be determined what structural elements are disrupted, how many times they are disrupted on average before the native state is reached, and whether the unfolding activity consists solely of unwinding RNA helices or whether RNA tertiary contacts are disrupted directly by DEAD-box proteins.
Group II introns
Group II introns are a second major class of autocatalytic introns.Citation129,Citation130 They splice themselves from precursor RNAs by a mechanism that involves the formation of an intron lariat RNA and is chemically identical to the splicing mechanism used by nuclear spliceosomal introns in higher organisms. The similarity in splicing mechanism and between the structures of group II intron domains and snRNAs suggest that group II introns were evolutionary ancestors of spliceosomal introns and the spliceosome.Citation129,Citation131 Indeed, some group II introns are mobile genetic elements and encode reverse transcriptases that function in both intron mobility and RNA splicing (“maturase” activity), explaining how group II introns may have initially spread within nuclear genomes before evolving into spliceosomal introns.Citation129 Thus, the roles played by DEAD-box proteins in splicing group II introns may provide insight into their roles in promoting RNP structural transitions in the spliceosome.Citation6,Citation88
To catalyze splicing, group II introns fold into a conserved tertiary structure in which six modular helical domains (DI-DVI) interact to form the RNA active site.Citation132,Citation133 DI is the largest domain and provides a scaffold for the assembly of other domains, including DV, which contains key nucleotide residues involved in catalysis, and DVI, which contains the branch-point adenosine. Like group I introns, the folding of group II introns is promoted by intramolecular RNA contacts and by proteins, which vary for different group II introns.Citation129 These proteins include intron-encoded reverse transcriptases with maturase activity (see above), host-encoded splicing factors that bind specifically to the intron RNA to stabilize the active RNA structure, and DEAD-box proteins.Citation22,Citation24,Citation129,Citation134,Citation135
Yeast mtDNA encodes four group II introns in the genes encoding cytochrome b (COB) and subunit I of cytochrome oxidase (COX1), and all four of these introns require Mss116 for efficient splicing in vivo.Citation22,Citation24 Two of these introns, aI1 and aI2, encode reverse transcriptase/maturases that function as intron-specific splicing factors to stabilize the active intron structure. In the absence of Mss116 in vivo, unspliced precursor RNAs containing aI2 accumulate in a complex with the intron-encoded maturase, indicating that Mss116 is not required for maturase binding and may function on the assembled or partially assembled RNP. Because CYT-19, which was known to function as an RNA chaperone in group I intron splicing, could replace Mss116 to promote the splicing of aI1 and aI2 in vivo, this later step was hypothesized to involve the resolution of kinetic traps.Citation24
The remaining two yeast mtDNA group II introns, aI5γ and bI1, do not encode maturases. A key step in analyzing the splicing of these introns was the development of in vitro systems in which purified DEAD-box proteins promote their splicing under near physiological conditions in vitro.Citation21,Citation136,Citation137 These in vitro systems better recapitulate biologically-relevant folding processes than the previous non-physiological conditions used to study these introns, and they have given important insights into the roles of DEAD-box proteins in group II intron folding. Mss116-promoted splicing of aI5γ and bI1 in vitro is strictly dependent upon ATP and is completely inhibited by motif I mutations that ablate ATP binding or hydrolysis.Citation128,Citation137,Citation138 Reports that the motif III mutant of Mss116 (SAT→AAA) could promote splicing of aI5γ without unwinding RNACitation137,Citation139 were refuted by showing that this mutant retains ATP-dependent RNA-unwinding activity for short duplexes and that this residual RNA-unwinding activity correlates with splicing activity in vitro and in vivo.Citation128,Citation138 Further, CYT-19 and Ded1 can replace Mss116 to promote the splicing of these introns in vitro and in vivo, indicating non-specific interactions,Citation21,Citation98,Citation136,Citation137 and the ability of different DEAD-box proteins to promote splicing in vitro and in vivo also correlates with their RNA-unwinding activity.Citation98,Citation138 Importantly, protease-digestion experiments showed that CYT-19 acts on bI1 as a classical RNA chaperone that promotes RNA folding in an ATP-dependent manner and is no longer required after RNA folding has occurred.Citation136 Although other DEAD-box protein activities may contribute to the splicing of some introns (see below), collectively, these findings indicate that Mss116 functions in splicing yeast mt group II introns as it does in splicing group I introns primarily by using its ATP-dependent RNA unwinding activity to function as a non-specific RNA chaperone.
The in vitro splicing systems have also given information on the kinetically-trapped structures by demonstrating the involvement of exon sequences. Early studies showed that the self-splicing rate of aI5γ is strongly decreased by inhibitory sequences in the 5′ exon, as found for group I introns (see above).Citation140 More recent studies of Mss116-promoted splicing of aI5γ in vitro have indicated that long exons do indeed contribute to kinetic traps, either by misfolding or by stabilizing inactive structures within the introns.Citation138,Citation139 Exon sequences are likely to make a substantial contribution to kinetic traps in vivo, where COX1 precursor RNAs have long and heterogenous 5′ exons, reflecting different orders in which introns upstream of aI5γ are spliced.Citation138
To probe further how DEAD-box proteins promote folding steps within the intron, further studies have used ribozyme constructs, analogous to those of group I introns, or constructs with very short exon sequences that minimize the potential for exons to contribute to kinetic traps. Early work indicated that under non-physiological conditions of high ionic strength and elevated temperature, a construct that includes domains I, III and V (denoted D135 RNA) folds to the native state on the minutes time scale without being rate-limited by kinetic traps.Citation141,Citation142 On the other hand, folding of this construct under near-physiological conditions is much slower and more complex, with multiple pathways and folding times extending to hours.Citation138,Citation143 An important advance in our understanding of how Mss116 promotes folding of aI5γ under these latter conditions came from a recent study in which a single molecule fluorescence approach was used to measure folding of a dual-labeled D135 ribozyme.Citation144 These experiments revealed that Mss116-promoted folding involves two discrete transitions that were detected by FRET between the two dyes: first there is an ATP-independent step, which most likely includes compaction of DI,Citation143,Citation145,Citation146 and then a second step that is ATP dependent. The authors favored the interpretation that this second step involves dissociation of bound Mss116 to permit further folding but left open the possibility of ATP-dependent RNA unwinding to resolve a kinetic trap. In a separate study, a two-stage assay that separates RNA folding from catalysis showed that maximal stimulation of folding requires ATP and that even when Mss116 is removed artificially by proteolysis, ATP-independent folding to the native state occurs only for a subpopulation of the D135 ribozyme, indicating multiple folding pathways.Citation138 Analogous experiments using a construct that included short exons indicated that removal of Mss116 by proteolysis does not result in detectable self-splicing,Citation138 suggesting that even short exons increase the probability of kinetic traps late in folding, either because they can participate in misfolding or because they promote and/or stabilize misfolding within the intron.
To probe in more detail the folding transitions in the presence and absence of Mss116, a recent study used nucleotide analog interference mapping (NAIM) with a native gel separation of compact and non-compact RNA, using constructs of aI5γ that include short exons.Citation147 The central findings were that a substantial number of modifications blocked or slowed compaction, with some of the effects larger in the presence of Mss116, and that most of the interferences resulted from modifications that would be expected to weaken base pairs. The authors interpreted the identities of the interfering nucleotides to indicate that the formation of base pairs is rate-limiting for folding, and they interpreted the larger interferences in the presence of Mss116 to indicate a greater importance of base-pair-forming steps in Mss116-mediated folding. Thus, they suggest that Mss116 promotes compaction of D1 by structural stabilization of an obligate intermediate and does not act as a helicase to unwind misfolded structures within the intron. Because these experiments monitored only the first transition – compaction – conclusions about other steps in intron folding are not warranted. In addition, the experiments in the absence of Mss116 monitored only the 15–20% of the ribozyme population that compacted within 45 min, and not the remaining 80–85%, the majority of which folds along other pathways involving additional steps that may be accelerated by Mss116.
Considered together, the simplest model that emerges from the data for aI5γ splicing is that Mss116 is needed to resolve kinetic traps involving the exon sequences and additionally facilitates folding steps within the intron. Mss116 promotes a major early intron folding step, compaction of D1, without requiring ATP. Upon dissociation of Mss116, one or more additional intermediates are encountered by most of the population for the D135 ribozyme and essentially the entire population for long or short exon-containing constructs. The intermediate(s) most likely includes non-native structures, which may include or be stabilized by exons when present, and Mss116 functions by using ATP-dependent RNA unwinding activity to promote partial unfolding and permit folding to the native state. Further work is needed to refine knowledge of the molecular events associated with the multiple folding pathways through this compaction step and the acceleration by Mss116 along these different pathways, as well as to probe the ATP-dependent steps that follow compaction and are required for folding of most of the intron population to the native state.
A final point is that although DEAD-box proteins dramatically accelerate splicing of the aI5γ and bI1 introns, the in vitro reactions are still relatively slow (2–4 x 10−2 min−1), suggesting that additional proteins might contribute in vivo and be necessary to reconstitute the complete physiological folding pathway for these introns in vitro.
Recent studies have shown that DEAD-box proteins also function in splicing of plant mt and chloroplast group II introns.Citation134,Citation135 The splicing of these plant group II introns involve large RNP complexes with multiple proteins, more analogous to the spliceosome. The roles of DEAD-box proteins in these more complex systems remain to be elucidated.
Future prospects
Although the progress in understanding the properties and roles of DEAD-box proteins has been substantial, many exciting questions remain unanswered. In terms of the biological roles, the full set of interactions with cellular RNAs is just beginning to be explored. Although DEAD-box proteins such as Mss116, CYT-19 and others like Ded1 and eIF4A do not have high specificity for individual RNA targets, it remains possible that they have preferred physiological targets. Conversely, DEAD-box proteins that are targeted to specific RNAs could still interact less specifically with other cellular RNAs. Recent studies indicate that the interactions of DEAD-box proteins and their effects on RNA structure begin during transcription. Mss116 has been found to interact with the mt RNA polymerase in vivo, positioning it to influence the folding of nascent RNAs.Citation148 The yeast protein Dbp2, the ortholog of human p68, was shown to associate with chromatin and prevent accumulation of cryptic, non-functional transcripts.Citation149 This activity requires the ability to bind and hydrolyze ATP, and together with known properties of DEAD-box proteins suggests that Dbp2 functions as a chaperone, promoting transient unfolding and rearrangements of these cryptic transcripts to permit their degradation. These and other results suggest that the roles of DEAD-box proteins as chaperones of RNA folding may be very broad, and they highlight the importance of further research toward a complete molecular understanding of these remarkable proteins.
| Abbreviations: | ||
| C-tail | = | C-terminal tail |
| CTE | = | C-terminal extension |
| dsDNA | = | double-stranded DNA |
| dsRNA | = | double-stranded RNA |
| DI-DVI | = | group II intron domains I-VI |
| FRET | = | fluorescence resonance energy transfer |
| mt | = | mitochondrial |
| NTE | = | N-terminal extension |
| SAXS | = | small angle X-ray scattering |
| ssRNA | = | single-stranded RNA |
Acknowledgments
This work was supported by grants from the NIH to R.R. (GM070456) and A.M.L. (GM037951) and by a grant by the Welch Foundation to R.R. (F-1563).
References
- Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 2010; 20:313 - 24; http://dx.doi.org/10.1016/j.sbi.2010.03.011; PMID: 20456941
- Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci 2011; 36:19 - 29; http://dx.doi.org/10.1016/j.tibs.2010.07.008; PMID: 20813532
- Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 2011; 12:505 - 16; http://dx.doi.org/10.1038/nrm3154; PMID: 21779027
- Noller HF. RNA structure: reading the ribosome. Science 2005; 309:1508 - 14; http://dx.doi.org/10.1126/science.1111771; PMID: 16141058
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009; 136:642 - 55; http://dx.doi.org/10.1016/j.cell.2009.01.035; PMID: 19239886
- Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136:701 - 18; http://dx.doi.org/10.1016/j.cell.2009.02.009; PMID: 19239890
- Collins K. The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol 2006; 7:484 - 94; http://dx.doi.org/10.1038/nrm1961; PMID: 16829980
- Esakova O, Krasilnikov AS. Of proteins and RNA: the RNase P/MRP family. RNA 2010; 16:1725 - 47; http://dx.doi.org/10.1261/rna.2214510; PMID: 20627997
- Hilbert M, Karow AR, Klostermeier D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol Chem 2009; 390:1237 - 50; http://dx.doi.org/10.1515/BC.2009.135; PMID: 19747077
- Jarmoskaite I, Russell R. DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip Rev RNA 2011; 2:135 - 52; http://dx.doi.org/10.1002/wrna.50; PMID: 21297876
- Pan C, Russell R. Roles of DEAD-box proteins in RNA and RNP Folding. RNA Biol 2010; 7:667 - 76; http://dx.doi.org/10.4161/rna.7.6.13571; PMID: 21045543
- Herschlag D. RNA chaperones and the RNA folding problem. J Biol Chem 1995; 270:20871 - 4; PMID: 7545662
- Schmid SR, Linder P. D-E-A-D protein family of putative RNA helicases. Mol Microbiol 1992; 6:283 - 91; http://dx.doi.org/10.1111/j.1365-2958.1992.tb01470.x; PMID: 1552844
- Lorsch JR, Herschlag D. The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry 1998; 37:2180 - 93; http://dx.doi.org/10.1021/bi972430g; PMID: 9485364
- Fairman ME, Maroney PA, Wang W, Bowers HA, Gollnick P, Nilsen TW, et al. Protein displacement by DExH/D “RNA helicases” without duplex unwinding. Science 2004; 304:730 - 4; http://dx.doi.org/10.1126/science.1095596; PMID: 15118161
- Jankowsky E, Bowers H. Remodeling of ribonucleoprotein complexes with DExH/D RNA helicases. Nucleic Acids Res 2006; 34:4181 - 8; http://dx.doi.org/10.1093/nar/gkl410; PMID: 16935886
- Tran EJ, Zhou Y, Corbett AH, Wente SR. The DEAD-box protein Dbp5 controls mRNA export by triggering specific RNA:protein remodeling events. Mol Cell 2007; 28:850 - 9; http://dx.doi.org/10.1016/j.molcel.2007.09.019; PMID: 18082609
- Rössler OG, Straka A, Stahl H. Rearrangement of structured RNA via branch migration structures catalysed by the highly related DEAD-box proteins p68 and p72. Nucleic Acids Res 2001; 29:2088 - 96; http://dx.doi.org/10.1093/nar/29.10.2088; PMID: 11353078
- Yang Q, Jankowsky E. ATP- and ADP-dependent modulation of RNA unwinding and strand annealing activities by the DEAD-box protein DED1. Biochemistry 2005; 44:13591 - 601; http://dx.doi.org/10.1021/bi0508946; PMID: 16216083
- Uhlmann-Schiffler H, Jalal C, Stahl H. Ddx42p--a human DEAD box protein with RNA chaperone activities. Nucleic Acids Res 2006; 34:10 - 22; http://dx.doi.org/10.1093/nar/gkj403; PMID: 16397294
- Halls C, Mohr S, Del Campo M, Yang Q, Jankowsky E, Lambowitz AM. Involvement of DEAD-box proteins in group I and group II intron splicing. Biochemical characterization of Mss116p, ATP hydrolysis-dependent and -independent mechanisms, and general RNA chaperone activity. J Mol Biol 2007; 365:835 - 55; http://dx.doi.org/10.1016/j.jmb.2006.09.083; PMID: 17081564
- Séraphin B, Simon M, Boulet A, Faye G. Mitochondrial splicing requires a protein from a novel helicase family. Nature 1989; 337:84 - 7; http://dx.doi.org/10.1038/337084a0; PMID: 2535893
- Mohr S, Stryker JM, Lambowitz AM. A DEAD-box protein functions as an ATP-dependent RNA chaperone in group I intron splicing. Cell 2002; 109:769 - 79; http://dx.doi.org/10.1016/S0092-8674(02)00771-7; PMID: 12086675
- Huang HR, Rowe CE, Mohr S, Jiang Y, Lambowitz AM, Perlman PS. The splicing of yeast mitochondrial group I and group II introns requires a DEAD-box protein with RNA chaperone function. Proc Natl Acad Sci USA 2005; 102:163 - 8; http://dx.doi.org/10.1073/pnas.0407896101; PMID: 15618406
- Caruthers JM, Johnson ER, McKay DB. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc Natl Acad Sci USA 2000; 97:13080 - 5; http://dx.doi.org/10.1073/pnas.97.24.13080; PMID: 11087862
- Story RM, Li H, Abelson JN. Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii.. Proc Natl Acad Sci USA 2001; 98:1465 - 70; http://dx.doi.org/10.1073/pnas.98.4.1465; PMID: 11171974
- Shi H, Cordin O, Minder CM, Linder P, Xu RM. Crystal structure of the human ATP-dependent splicing and export factor UAP56. Proc Natl Acad Sci USA 2004; 101:17628 - 33; http://dx.doi.org/10.1073/pnas.0408172101; PMID: 15585580
- Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, et al. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 2006; 313:1968 - 72; http://dx.doi.org/10.1126/science.1131981; PMID: 16931718
- Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 2006; 125:287 - 300; http://dx.doi.org/10.1016/j.cell.2006.01.054; PMID: 16630817
- Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 2006; 126:713 - 25; http://dx.doi.org/10.1016/j.cell.2006.08.006; PMID: 16923391
- Del Campo M, Lambowitz AM. Structure of the Yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol Cell 2009; 35:598 - 609; http://dx.doi.org/10.1016/j.molcel.2009.07.032; PMID: 19748356
- Fan JS, Cheng Z, Zhang J, Noble C, Zhou Z, Song H, et al. Solution and crystal structures of mRNA exporter Dbp5p and its interaction with nucleotides. J Mol Biol 2009; 388:1 - 10; http://dx.doi.org/10.1016/j.jmb.2009.03.004; PMID: 19281819
- Nielsen KH, Chamieh H, Andersen CB, Fredslund F, Hamborg K, Le Hir H, et al. Mechanism of ATP turnover inhibition in the EJC. RNA 2009; 15:67 - 75; http://dx.doi.org/10.1261/rna.1283109; PMID: 19033377
- Collins R, Karlberg T, Lehtiö L, Schütz P, van den Berg S, Dahlgren LG, et al. The DEXD/H-box RNA helicase DDX19 is regulated by an alpha-helical switch. J Biol Chem 2009; 284:10296 - 300; http://dx.doi.org/10.1074/jbc.C900018200; PMID: 19244245
- von Moeller H, Basquin C, Conti E. The mRNA export protein DBP5 binds RNA and the cytoplasmic nucleoporin NUP214 in a mutually exclusive manner. Nat Struct Mol Biol 2009; 16:247 - 54; http://dx.doi.org/10.1038/nsmb.1561; PMID: 19219046
- Tanner NK, Cordin O, Banroques J, Doère M, Linder P. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol Cell 2003; 11:127 - 38; http://dx.doi.org/10.1016/S1097-2765(03)00006-6; PMID: 12535527
- Cordin O, Tanner NK, Doère M, Linder P, Banroques J. The newly discovered Q motif of DEAD-box RNA helicases regulates RNA-binding and helicase activity. EMBO J 2004; 23:2478 - 87; http://dx.doi.org/10.1038/sj.emboj.7600272; PMID: 15201868
- Yang Q, Del Campo M, Lambowitz AM, Jankowsky E. DEAD-box proteins unwind duplexes by local strand separation. Mol Cell 2007; 28:253 - 63; http://dx.doi.org/10.1016/j.molcel.2007.08.016; PMID: 17964264
- Chen Y, Potratz JP, Tijerina P, Del Campo M, Lambowitz AM, Russell R. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc Natl Acad Sci USA 2008; 105:20203 - 8; http://dx.doi.org/10.1073/pnas.0811075106; PMID: 19088196
- Liu F, Putnam A, Jankowsky E. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc Natl Acad Sci USA 2008; 105:20209 - 14; http://dx.doi.org/10.1073/pnas.0811115106; PMID: 19088201
- Mohr G, Del Campo M, Turner KG, Gilman B, Wolf RZ, Lambowitz AM. High-throughput genetic identification of functionally important regions of the yeast DEAD-box protein Mss116p. J Mol Biol 2011; 413:952 - 72; http://dx.doi.org/10.1016/j.jmb.2011.09.015; PMID: 21945532
- Fuller-Pace FV, Nicol SM, Reid AD, Lane DP. DbpA: a DEAD box protein specifically activated by 23s rRNA. EMBO J 1993; 12:3619 - 26; PMID: 8253085
- Diges CM, Uhlenbeck OC. Escherichia coli DbpA is an RNA helicase that requires hairpin 92 of 23S rRNA. EMBO J 2001; 20:5503 - 12; http://dx.doi.org/10.1093/emboj/20.19.5503; PMID: 11574482
- Hardin JW, Hu YX, McKay DB. Structure of the RNA binding domain of a DEAD-box helicase bound to its ribosomal RNA target reveals a novel mode of recognition by an RNA recognition motif. J Mol Biol 2010; 402:412 - 27; http://dx.doi.org/10.1016/j.jmb.2010.07.040; PMID: 20673833
- Klostermeier D, Rudolph MG. A novel dimerization motif in the C-terminal domain of the Thermus thermophilus DEAD box helicase Hera confers substantial flexibility. Nucleic Acids Res 2009; 37:421 - 30; http://dx.doi.org/10.1093/nar/gkn947; PMID: 19050012
- Chimnaronk S, Suzuki T, Manita T, Ikeuchi Y, Yao M, Suzuki T, et al. RNA helicase module in an acetyltransferase that modifies a specific tRNA anticodon. EMBO J 2009; 28:1362 - 73; http://dx.doi.org/10.1038/emboj.2009.69; PMID: 19322199
- Lau PW, Guiley KZ, De N, Potter CS, Carragher B, MacRae IJ. The molecular architecture of human Dicer. Nat Struct Mol Biol 2012; 19:436 - 40; http://dx.doi.org/10.1038/nsmb.2268; PMID: 22426548
- Mohr G, Del Campo M, Mohr S, Yang Q, Jia H, Jankowsky E, et al. Function of the C-terminal domain of the DEAD-box protein Mss116p analyzed in vivo and in vitro.. J Mol Biol 2008; 375:1344 - 64; http://dx.doi.org/10.1016/j.jmb.2007.11.041; PMID: 18096186
- Grohman JK, Del Campo M, Bhaskaran H, Tijerina P, Lambowitz AM, Russell R. Probing the mechanisms of DEAD-box proteins as general RNA chaperones: the C-terminal domain of CYT-19 mediates general recognition of RNA. Biochemistry 2007; 46:3013 - 22; http://dx.doi.org/10.1021/bi0619472; PMID: 17311413
- Polach KJ, Uhlenbeck OC. Cooperative binding of ATP and RNA substrates to the DEAD/H protein DbpA. Biochemistry 2002; 41:3693 - 702; http://dx.doi.org/10.1021/bi012062n; PMID: 11888286
- Iost I, Dreyfus M, Linder P. Ded1p, a DEAD-box protein required for translation initiation in Saccharomyces cerevisiae, is an RNA helicase. J Biol Chem 1999; 274:17677 - 83; http://dx.doi.org/10.1074/jbc.274.25.17677; PMID: 10364207
- Banroques J, Cordin O, Doère M, Linder P, Tanner NK. A conserved phenylalanine of motif IV in superfamily 2 helicases is required for cooperative, ATP-dependent binding of RNA substrates in DEAD-box proteins. Mol Cell Biol 2008; 28:3359 - 71; http://dx.doi.org/10.1128/MCB.01555-07; PMID: 18332124
- Theissen B, Karow AR, Köhler J, Gubaev A, Klostermeier D. Cooperative binding of ATP and RNA induces a closed conformation in a DEAD box RNA helicase. Proc Natl Acad Sci USA 2008; 105:548 - 53; http://dx.doi.org/10.1073/pnas.0705488105; PMID: 18184816
- Aregger R, Klostermeier D. The DEAD box helicase YxiN maintains a closed conformation during ATP hydrolysis. Biochemistry 2009; 48:10679 - 81; http://dx.doi.org/10.1021/bi901278p; PMID: 19839642
- Henn A, Cao W, Hackney DD, De La Cruz EM. The ATPase cycle mechanism of the DEAD-box rRNA helicase, DbpA. J Mol Biol 2008; 377:193 - 205; http://dx.doi.org/10.1016/j.jmb.2007.12.046; PMID: 18237742
- Cao W, Coman MM, Ding S, Henn A, Middleton ER, Bradley MJ, et al. Mechanism of Mss116 ATPase reveals functional diversity of DEAD-box proteins. J Mol Biol 2011; 409:399 - 414; http://dx.doi.org/10.1016/j.jmb.2011.04.004; PMID: 21501623
- Mallam AL, Jarmoskaite I, Tijerina P, Del Campo M, Seifert S, Guo L, et al. Solution structures of DEAD-box RNA chaperones reveal conformational changes and nucleic acid tethering by a basic tail. Proc Natl Acad Sci USA 2011; 108:12254 - 9; http://dx.doi.org/10.1073/pnas.1109566108; PMID: 21746911
- Karow AR, Klostermeier D. A conformational change in the helicase core is necessary but not sufficient for RNA unwinding by the DEAD box helicase YxiN. Nucleic Acids Res 2009; 37:4464 - 71; http://dx.doi.org/10.1093/nar/gkp397; PMID: 19474341
- Banroques J, Doère M, Dreyfus M, Linder P, Tanner NK. Motif III in superfamily 2 “helicases” helps convert the binding energy of ATP into a high-affinity RNA binding site in the yeast DEAD-box protein Ded1. J Mol Biol 2010; 396:949 - 66; http://dx.doi.org/10.1016/j.jmb.2009.12.025; PMID: 20026132
- Tarn WY, Chang TH. The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA Biol 2009; 6:17 - 20; http://dx.doi.org/10.4161/rna.6.1.7440; PMID: 19106629
- Karow AR, Theissen B, Klostermeier D. Authentic interdomain communication in an RNA helicase reconstituted by expressed protein ligation of two helicase domains. FEBS J 2007; 274:463 - 73; http://dx.doi.org/10.1111/j.1742-4658.2006.05593.x; PMID: 17229151
- Rogers GW Jr., Richter NJ, Lima WF, Merrick WC. Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J Biol Chem 2001; 276:30914 - 22; http://dx.doi.org/10.1074/jbc.M100157200; PMID: 11418588
- Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, et al. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell 2009; 136:447 - 60; http://dx.doi.org/10.1016/j.cell.2009.01.014; PMID: 19203580
- Hilbert M, Kebbel F, Gubaev A, Klostermeier D. eIF4G stimulates the activity of the DEAD box protein eIF4A by a conformational guidance mechanism. Nucleic Acids Res 2011; 39:2260 - 70; http://dx.doi.org/10.1093/nar/gkq1127; PMID: 21062831
- Özeş AR, Feoktistova K, Avanzino BC, Fraser CS. Duplex unwinding and ATPase activities of the DEAD-box helicase eIF4A are coupled by eIF4G and eIF4B. J Mol Biol 2011; 412:674 - 87; http://dx.doi.org/10.1016/j.jmb.2011.08.004; PMID: 21840318
- Rajagopal V, Park EH, Hinnebusch AG, Lorsch JR. Specific domains in yeast translation initiation factor eIF4G strongly bias RNA unwinding activity of the eIF4F complex toward duplexes with 5′-overhangs. J Biol Chem 2012; 287:20301 - 12; http://dx.doi.org/10.1074/jbc.M112.347278; PMID: 22467875
- Schütz P, Bumann M, Oberholzer AE, Bieniossek C, Trachsel H, Altmann M, et al. Crystal structure of the yeast eIF4A-eIF4G complex: an RNA-helicase controlled by protein-protein interactions. Proc Natl Acad Sci USA 2008; 105:9564 - 9; http://dx.doi.org/10.1073/pnas.0800418105; PMID: 18606994
- Hilliker A, Gao Z, Jankowsky E, Parker R. The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol Cell 2011; 43:962 - 72; http://dx.doi.org/10.1016/j.molcel.2011.08.008; PMID: 21925384
- Bono F, Gehring NH. Assembly, disassembly and recycling: the dynamics of exon junction complexes. RNA Biol 2011; 8:24 - 9; http://dx.doi.org/10.4161/rna.8.1.13618; PMID: 21289489
- Gehring NH, Lamprinaki S, Kulozik AE, Hentze MW. Disassembly of exon junction complexes by PYM. Cell 2009; 137:536 - 48; http://dx.doi.org/10.1016/j.cell.2009.02.042; PMID: 19410547
- Lund MK, Guthrie C. The DEAD-box protein Dbp5p is required to dissociate Mex67p from exported mRNPs at the nuclear rim. Mol Cell 2005; 20:645 - 51; http://dx.doi.org/10.1016/j.molcel.2005.10.005; PMID: 16307927
- Ledoux S, Guthrie C. Regulation of the Dbp5 ATPase cycle in mRNP remodeling at the nuclear pore: a lively new paradigm for DEAD-box proteins. Genes Dev 2011; 25:1109 - 14; http://dx.doi.org/10.1101/gad.2062611; PMID: 21632821
- Valkov E, Dean JC, Jani D, Kuhlmann SI, Stewart M. Structural basis for the assembly and disassembly of mRNA nuclear export complexes. Biochim Biophys Acta 2012; 1819:578 - 92; http://dx.doi.org/10.1016/j.bbagrm.2012.02.017; PMID: 22406340
- Noble KN, Tran EJ, Alcázar-Román AR, Hodge CA, Cole CN, Wente SR. The Dbp5 cycle at the nuclear pore complex during mRNA export II: nucleotide cycling and mRNP remodeling by Dbp5 are controlled by Nup159 and Gle1. Genes Dev 2011; 25:1065 - 77; http://dx.doi.org/10.1101/gad.2040611; PMID: 21576266
- Montpetit B, Thomsen ND, Helmke KJ, Seeliger MA, Berger JM, Weis K. A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature 2011; 472:238 - 42; http://dx.doi.org/10.1038/nature09862; PMID: 21441902
- Yang Q, Jankowsky E. The DEAD-box protein Ded1 unwinds RNA duplexes by a mode distinct from translocating helicases. Nat Struct Mol Biol 2006; 13:981 - 6; http://dx.doi.org/10.1038/nsmb1165; PMID: 17072313
- Rogers GW Jr., Lima WF, Merrick WC. Further characterization of the helicase activity of eIF4A. Substrate specificity. J Biol Chem 2001; 276:12598 - 608; http://dx.doi.org/10.1074/jbc.M007560200; PMID: 11278350
- Huang Y, Liu ZR. The ATPase, RNA unwinding, and RNA binding activities of recombinant p68 RNA helicase. J Biol Chem 2002; 277:12810 - 5; http://dx.doi.org/10.1074/jbc.M200182200; PMID: 11823473
- Bizebard T, Ferlenghi I, Iost I, Dreyfus M. Studies on three E. coli DEAD-box helicases point to an unwinding mechanism different from that of model DNA helicases. Biochemistry 2004; 43:7857 - 66; http://dx.doi.org/10.1021/bi049852s; PMID: 15196029
- Henn A, Cao W, Licciardello N, Heitkamp SE, Hackney DD, De La Cruz EM. Pathway of ATP utilization and duplex rRNA unwinding by the DEAD-box helicase, DbpA. Proc Natl Acad Sci USA 2010; 107:4046 - 50; http://dx.doi.org/10.1073/pnas.0913081107; PMID: 20160110
- Wang Q, Arnold JJ, Uchida A, Raney KD, Cameron CE. Phosphate release contributes to the rate-limiting step for unwinding by an RNA helicase. Nucleic Acids Res 2010; 38:1312 - 24; http://dx.doi.org/10.1093/nar/gkp1118; PMID: 19969541
- Mallam AL, Del Campo M, Gilman B, Sidote DJ, Lambowitz AM. Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p. Nature 2012; 490:121 - 5; http://dx.doi.org/10.1038/nature11402; PMID: 22940866
- Tijerina P, Bhaskaran H, Russell R. Nonspecific binding to structured RNA and preferential unwinding of an exposed helix by the CYT-19 protein, a DEAD-box RNA chaperone. Proc Natl Acad Sci USA 2006; 103:16698 - 703; http://dx.doi.org/10.1073/pnas.0603127103; PMID: 17075070
- Kossen K, Karginov FV, Uhlenbeck OC. The carboxy-terminal domain of the DExDH protein YxiN is sufficient to confer specificity for 23S rRNA. J Mol Biol 2002; 324:625 - 36; http://dx.doi.org/10.1016/S0022-2836(02)01140-3; PMID: 12460566
- Wang S, Hu Y, Overgaard MT, Karginov FV, Uhlenbeck OC, McKay DB. The domain of the Bacillus subtilis DEAD-box helicase YxiN that is responsible for specific binding of 23S rRNA has an RNA recognition motif fold. RNA 2006; 12:959 - 67; http://dx.doi.org/10.1261/rna.5906; PMID: 16611943
- Wang S, Overgaard MT, Hu Y, McKay DB. The Bacillus subtilis RNA helicase YxiN is distended in solution. Biophys J 2008; 94:L01 - 03; http://dx.doi.org/10.1529/biophysj.107.120709; PMID: 17951299
- Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 1998; 92:315 - 26; http://dx.doi.org/10.1016/S0092-8674(00)80925-3; PMID: 9476892
- Stevens SW. (2010). The biology of DEAH/RHA proteins and their mechanism of action. In RNA helicases (Jankowsky, E., ed.), Vol. RSC Biomolecular Sciences No. 19, pp. 99-120. Royal Society of Chemistry.
- Strunk BS, Karbstein K. Powering through ribosome assembly. RNA 2009; 15:2083 - 104; http://dx.doi.org/10.1261/rna.1792109; PMID: 19850913
- Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Annu Rev Biochem 2011; 80:501 - 26; http://dx.doi.org/10.1146/annurev-biochem-062608-160432; PMID: 21529161
- Russell R. RNA misfolding and the action of chaperones. Front Biosci 2008; 13:1 - 20; http://dx.doi.org/10.2741/2557; PMID: 17981525
- Cech TR. Self-splicing of group I introns. Annu Rev Biochem 1990; 59:543 - 68; http://dx.doi.org/10.1146/annurev.bi.59.070190.002551; PMID: 2197983
- Michel F, Westhof E. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J Mol Biol 1990; 216:585 - 610; http://dx.doi.org/10.1016/0022-2836(90)90386-Z; PMID: 2258934
- Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science 1996; 273:1678 - 85; http://dx.doi.org/10.1126/science.273.5282.1678; PMID: 8781224
- Golden BL, Gooding AR, Podell ER, Cech TR. A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science 1998; 282:259 - 64; http://dx.doi.org/10.1126/science.282.5387.259; PMID: 9841391
- Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature 2004; 430:45 - 50; http://dx.doi.org/10.1038/nature02642; PMID: 15175762
- Lambowitz AM, Perlman PS. Involvement of aminoacyl-tRNA synthetases and other proteins in group I and group II intron splicing. Trends Biochem Sci 1990; 15:440 - 4; http://dx.doi.org/10.1016/0968-0004(90)90283-H; PMID: 2278103
- Del Campo M, Mohr S, Jiang Y, Jia H, Jankowsky E, Lambowitz AM. Unwinding by local strand separation is critical for the function of DEAD-box proteins as RNA chaperones. J Mol Biol 2009; 389:674 - 93; http://dx.doi.org/10.1016/j.jmb.2009.04.043; PMID: 19393667
- Bifano AL, Caprara MG. A DExH/D-box protein coordinates the two steps of splicing in a group I intron. J Mol Biol 2008; 383:667 - 82; http://dx.doi.org/10.1016/j.jmb.2008.08.070; PMID: 18789947
- Lindahl T, Adams A, Fresco JR. Renaturation of transfer ribonucleic acids through site binding of magnesium. Proc Natl Acad Sci USA 1966; 55:941 - 8; http://dx.doi.org/10.1073/pnas.55.4.941; PMID: 5327073
- Aubert M, Scott JF, Reynier M, Monier R. Rearrangement of the conformation of Escherichia coli 5S RNA. Proc Natl Acad Sci USA 1968; 61:292 - 9; http://dx.doi.org/10.1073/pnas.61.1.292; PMID: 4880610
- Walstrum SA, Uhlenbeck OC. The self-splicing RNA of Tetrahymena is trapped in a less active conformation by gel purification. Biochemistry 1990; 29:10573 - 6; http://dx.doi.org/10.1021/bi00498a022; PMID: 2271667
- Woodson SA, Cech TR. Alternative secondary structures in the 5′ exon affect both forward and reverse self-splicing of the Tetrahymena intervening sequence RNA. Biochemistry 1991; 30:2042 - 50; http://dx.doi.org/10.1021/bi00222a006; PMID: 1998665
- Pan J, Woodson SA. Folding intermediates of a self-splicing RNA: mispairing of the catalytic core. J Mol Biol 1998; 280:597 - 609; http://dx.doi.org/10.1006/jmbi.1998.1901; PMID: 9677291
- Russell R, Herschlag D. New pathways in folding of the Tetrahymena group I RNA enzyme. J Mol Biol 1999; 291:1155 - 67; http://dx.doi.org/10.1006/jmbi.1999.3026; PMID: 10518951
- Zhang L, Bao P, Leibowitz MJ, Zhang Y. Slow formation of a pseudoknot structure is rate limiting in the productive co-transcriptional folding of the self-splicing Candida intron. RNA 2009; 15:1986 - 92; http://dx.doi.org/10.1261/rna.1638609; PMID: 19710184
- Duncan CD, Weeks KM. SHAPE analysis of long-range interactions reveals extensive and thermodynamically preferred misfolding in a fragile group I intron RNA. Biochemistry 2008; 47:8504 - 13; http://dx.doi.org/10.1021/bi800207b; PMID: 18642882
- Pan T, Sosnick TR. Intermediates and kinetic traps in the folding of a large ribozyme revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity. Nat Struct Biol 1997; 4:931 - 8; http://dx.doi.org/10.1038/nsb1197-931; PMID: 9360610
- Chadalavada DM, Senchak SE, Bevilacqua PC. The folding pathway of the genomic hepatitis delta virus ribozyme is dominated by slow folding of the pseudoknots. J Mol Biol 2002; 317:559 - 75; http://dx.doi.org/10.1006/jmbi.2002.5434; PMID: 11955009
- Treiber DK, Williamson JR. Exposing the kinetic traps in RNA folding. Curr Opin Struct Biol 1999; 9:339 - 45; http://dx.doi.org/10.1016/S0959-440X(99)80045-1; PMID: 10361090
- Rangan P, Masquida B, Westhof E, Woodson SA. Assembly of core helices and rapid tertiary folding of a small bacterial group I ribozyme. Proc Natl Acad Sci USA 2003; 100:1574 - 9; http://dx.doi.org/10.1073/pnas.0337743100; PMID: 12574513
- Roh JH, Guo L, Kilburn JD, Briber RM, Irving T, Woodson SA. Multistage collapse of a bacterial ribozyme observed by time-resolved small-angle X-ray scattering. J Am Chem Soc 2010; 132:10148 - 54; http://dx.doi.org/10.1021/ja103867p; PMID: 20597502
- Sinan S, Yuan X, Russell R. The Azoarcus group I intron ribozyme misfolds and is accelerated for refolding by ATP-dependent RNA chaperone proteins. J Biol Chem 2011; 286:37304 - 12; http://dx.doi.org/10.1074/jbc.M111.287706; PMID: 21878649
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena.. Cell 1982; 31:147 - 57; http://dx.doi.org/10.1016/0092-8674(82)90414-7; PMID: 6297745
- Woodson SA, Emerick VL. An alternative helix in the 26S rRNA promotes excision and integration of the Tetrahymena intervening sequence. Mol Cell Biol 1993; 13:1137 - 45; PMID: 8380892
- Emerick VL, Woodson SA. Fingerprinting the folding of a group I precursor RNA. Proc Natl Acad Sci USA 1994; 91:9675 - 9; http://dx.doi.org/10.1073/pnas.91.21.9675; PMID: 7937871
- Zaug AJ, Grosshans CA, Cech TR. Sequence-specific endoribonuclease activity of the Tetrahymena ribozyme: enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme-substrate complexes. Biochemistry 1988; 27:8924 - 31; http://dx.doi.org/10.1021/bi00425a008; PMID: 3069131
- Wan Y, Mitchell D 3rd, Russell R. Catalytic activity as a probe of native RNA folding. Methods Enzymol 2009; 468:195 - 218; http://dx.doi.org/10.1016/S0076-6879(09)68010-1; PMID: 20946771
- Potratz JP, Russell R. RNA catalysis as a probe for chaperone activity of DEAD-box helicases. Methods Enzymol 2012; 511:111 - 30; PMID: 22713317
- Zarrinkar PP, Williamson JR. Kinetic intermediates in RNA folding. Science 1994; 265:918 - 24; http://dx.doi.org/10.1126/science.8052848; PMID: 8052848
- Sclavi B, Sullivan M, Chance MR, Brenowitz M, Woodson SA. RNA folding at millisecond intervals by synchrotron hydroxyl radical footprinting. Science 1998; 279:1940 - 3; http://dx.doi.org/10.1126/science.279.5358.1940; PMID: 9506944
- Russell R, Herschlag D. Probing the folding landscape of the Tetrahymena ribozyme: commitment to form the native conformation is late in the folding pathway. J Mol Biol 2001; 308:839 - 51; http://dx.doi.org/10.1006/jmbi.2001.4751; PMID: 11352576
- Russell R, Zhuang X, Babcock HP, Millett IS, Doniach S, Chu S, et al. Exploring the folding landscape of a structured RNA. Proc Natl Acad Sci USA 2002; 99:155 - 60; http://dx.doi.org/10.1073/pnas.221593598; PMID: 11756689
- Laederach A, Shcherbakova I, Jonikas MA, Altman RB, Brenowitz M. Distinct contribution of electrostatics, initial conformational ensemble, and macromolecular stability in RNA folding. Proc Natl Acad Sci USA 2007; 104:7045 - 50; http://dx.doi.org/10.1073/pnas.0608765104; PMID: 17438287
- Shcherbakova I, Mitra S, Laederach A, Brenowitz M. Energy barriers, pathways, and dynamics during folding of large, multidomain RNAs. Curr Opin Chem Biol 2008; 12:655 - 66; http://dx.doi.org/10.1016/j.cbpa.2008.09.017; PMID: 18926923
- Russell R, Das R, Suh H, Travers KJ, Laederach A, Engelhardt MA, et al. The paradoxical behavior of a highly structured misfolded intermediate in RNA folding. J Mol Biol 2006; 363:531 - 44; http://dx.doi.org/10.1016/j.jmb.2006.08.024; PMID: 16963081
- Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature 2007; 449:1014 - 8; http://dx.doi.org/10.1038/nature06235; PMID: 17960235
- Del Campo M, Tijerina P, Bhaskaran H, Mohr S, Yang Q, Jankowsky E, et al. Do DEAD-box proteins promote group II intron splicing without unwinding RNA?. Mol Cell 2007; 28:159 - 66; http://dx.doi.org/10.1016/j.molcel.2007.07.028; PMID: 17936712
- Lambowitz AM, Zimmerly S. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol 2011; 3:a003616; http://dx.doi.org/10.1101/cshperspect.a003616; PMID: 20463000
- Pyle AM. The tertiary structure of group II introns: implications for biological function and evolution. Crit Rev Biochem Mol Biol 2010; 45:215 - 32; http://dx.doi.org/10.3109/10409231003796523; PMID: 20446804
- Keating KS, Toor N, Perlman PS, Pyle AM. A structural analysis of the group II intron active site and implications for the spliceosome. RNA 2010; 16:1 - 9; http://dx.doi.org/10.1261/rna.1791310; PMID: 19948765
- Dai L, Chai D, Gu SQ, Gabel J, Noskov SY, Blocker FJ, et al. A three-dimensional model of a group II intron RNA and its interaction with the intron-encoded reverse transcriptase. Mol Cell 2008; 30:472 - 85; http://dx.doi.org/10.1016/j.molcel.2008.04.001; PMID: 18424209
- Toor N, Keating KS, Fedorova O, Rajashankar K, Wang J, Pyle AM. Tertiary architecture of the Oceanobacillus iheyensis group II intron. RNA 2010; 16:57 - 69; http://dx.doi.org/10.1261/rna.1844010; PMID: 19952115
- Köhler D, Schmidt-Gattung S, Binder S. The DEAD-box protein PMH2 is required for efficient group II intron splicing in mitochondria of Arabidopsis thaliana.. Plant Mol Biol 2010; 72:459 - 67; http://dx.doi.org/10.1007/s11103-009-9584-9; PMID: 19960362
- Asakura Y, Galarneau E, Watkins KP, Barkan A, van Wijk KJ. Chloroplast RH3 DEAD box RNA helicases in maize and arabidopsis function in splicing of specific group II introns and affect chloroplast ribosome biogenesis. Plant Physiol 2012; 159:961 - 74; http://dx.doi.org/10.1104/pp.112.197525; PMID: 22576849
- Mohr S, Matsuura M, Perlman PS, Lambowitz AM. A DEAD-box protein alone promotes group II intron splicing and reverse splicing by acting as an RNA chaperone. Proc Natl Acad Sci USA 2006; 103:3569 - 74; http://dx.doi.org/10.1073/pnas.0600332103; PMID: 16505350
- Solem A, Zingler N, Pyle AM. A DEAD protein that activates intron self-splicing without unwinding RNA. Mol Cell 2006; 24:611 - 7; http://dx.doi.org/10.1016/j.molcel.2006.10.032; PMID: 17188036
- Potratz JP, Del Campo M, Wolf RZ, Lambowitz AM, Russell R. ATP-dependent roles of the DEAD-box protein Mss116p in group II intron splicing in vitro and in vivo. J Mol Biol 2011; 411:661 - 79; http://dx.doi.org/10.1016/j.jmb.2011.05.047; PMID: 21679717
- Zingler N, Solem A, Pyle AM. Dual roles for the Mss116 cofactor during splicing of the ai5γ group II intron. Nucleic Acids Res 2010; 38:6602 - 9; http://dx.doi.org/10.1093/nar/gkq530; PMID: 20554854
- Nolte A, Chanfreau G, Jacquier A. Influence of substrate structure on in vitro ribozyme activity of a group II intron. RNA 1998; 4:694 - 708; http://dx.doi.org/10.1017/S1355838298980165; PMID: 9622128
- Swisher JF, Su LJ, Brenowitz M, Anderson VE, Pyle AM. Productive folding to the native state by a group II intron ribozyme. J Mol Biol 2002; 315:297 - 310; http://dx.doi.org/10.1006/jmbi.2001.5233; PMID: 11786013
- Su LJ, Brenowitz M, Pyle AM. An alternative route for the folding of large RNAs: apparent two-state folding by a group II intron ribozyme. J Mol Biol 2003; 334:639 - 52; http://dx.doi.org/10.1016/j.jmb.2003.09.071; PMID: 14636593
- Fedorova O, Solem A, Pyle AM. Protein-facilitated folding of group II intron ribozymes. J Mol Biol 2010; 397:799 - 813; http://dx.doi.org/10.1016/j.jmb.2010.02.001; PMID: 20138894
- Karunatilaka KS, Solem A, Pyle AM, Rueda D. Single-molecule analysis of Mss116-mediated group II intron folding. Nature 2010; 467:935 - 9; http://dx.doi.org/10.1038/nature09422; PMID: 20944626
- Waldsich C, Pyle AM. A folding control element for tertiary collapse of a group II intron ribozyme. Nat Struct Mol Biol 2007; 14:37 - 44; http://dx.doi.org/10.1038/nsmb1181; PMID: 17143279
- Waldsich C, Pyle AM. A kinetic intermediate that regulates proper folding of a group II intron RNA. J Mol Biol 2008; 375:572 - 80; http://dx.doi.org/10.1016/j.jmb.2007.10.052; PMID: 18022197
- Fedorova O, Pyle AM. The brace for a growing scaffold: mss116 protein promotes RNA folding by stabilizing an early assembly intermediate. J Mol Biol 2012; 422:347 - 65; http://dx.doi.org/10.1016/j.jmb.2012.05.037; PMID: 22705286
- Markov DA, Savkina M, Anikin M, Del Campo M, Ecker K, Lambowitz AM, et al. Identification of proteins associated with the yeast mitochondrial RNA polymerase by tandem affinity purification. Yeast 2009; 26:423 - 40; http://dx.doi.org/10.1002/yea.1672; PMID: 19536766
- Cloutier SC, Ma WK, Nguyen LT, Tran EJ. The DEAD-box RNA helicase Dbp2 connects RNA quality control with repression of aberrant transcription. J Biol Chem 2012; 287:26155 - 66; http://dx.doi.org/10.1074/jbc.M112.383075; PMID: 22679025
- Lehnert V, Jaeger L, Michel F, Westhof E. New loop-loop tertiary interactions in self-splicing introns of subgroup IC and ID: a complete 3D model of the Tetrahymena thermophila ribozyme. Chem Biol 1996; 3:993 - 1009; http://dx.doi.org/10.1016/S1074-5521(96)90166-0; PMID: 9000010