
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Theoretical studies were carried out to investigate the thermal decomposition of ethyl dithiocarbonates (xanthate) using Hartree–Fock at the HF/321-G* level and the density functional method with Becke 3 Lee Yang pair DFT/(B3LYP), 6-31G*, 6-31G**, 6-31+G*, MP2 and CCSD in the ab initio method of calculation using Spartan 10. Geometric parameters, such as the bond length, bond angles, dihedral angles, heat of formation, atomic charges and vibrational frequencies, were obtained. The data were used to calculate the thermodynamics parameters, change in entropy ΔS, enthalpy change ΔH, free energy G, pre-exponential factor A, rate k at 623 K, and variation of rate k with temperatures from 498–623 K at temperature intervals of 25 K. It was observed that the values obtained are in good agreement with the experimental values for the ab initio methods, and according to Arrhenius theory, the calculated rate k increases with increasing temperature.
1 Introduction
Pyrolysis of dithiocarbonates (xanthates) and carbonate related compounds are valuable routes to synthesizing alkenes without rearranging the carbon skeleton. Pyrolysis of xanthate precursors was first reported by Chugaev in 1899 in studies on the optical properties of xanthates [Citation1].
Table 2 Selected bond angles in degrees for the pyrolysis of O-ethyl, S-methyl xanthate (dithiocarbonates).
Table 3 Selected dihedral angles in degrees for the pyrolysis of O-ethyl S-methyl xanthate (dithiocarbonate).
The preparation of xanthates was carried out by reacting the corresponding alcohol with a base and carbon disulphide, accompanied by an alkyl iodide (usually methyl iodide) [Citation14]. For pyrolysis to produce alkenes, together with the elimination of by-products (carbonyl sulphide and a thiol), β-hydrogen is required, and when more than one β-hydrogen is present, regioselectivity has to be considered, which limits the Chugaev reaction. Xanthates with no β-hydrogen will undergo thione-to-thiol rearrangements, giving S,S-dialkyl dithiocarbonates. The mechanism of the Chugaev reaction was proposed to involve two possible pathways. The first pathway reacts in a one-step mechanism, yielding an alkene, carbonyl sulphide and thiol. In the second mechanism, a thione group attacking a β-hydrogen yields an alkene and an unstable S-alkyl dithiocarbonate intermediate, which will decompose to carbonyl sulphide and a thiol ().
Scheme 1 Two proposed pathways for the pyrolysis of xanthates.
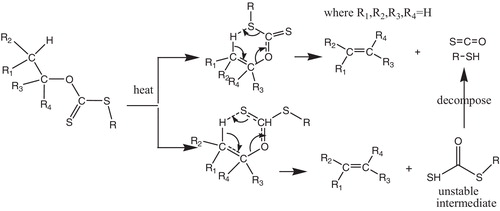
The first pathway was supported by Huckel et al. [Citation2], while Barton and Cram [Citation3,Citation4] preferred the second mechanism. Experimental studies were also applied to investigate the Chugaev mechanisms. Alexander and Mudrak [Citation5,Citation6] demonstrated the cis-elimination mechanism; Bader and Bourns [Citation7] conducted isotope effect studies for the pyrolysis of trans-2-methyl-1-indanyl xanthate and found solid evidence for the second pathway; further kinetic and thermodynamic studies concluded that the Chugaev reactions were homogenous and unimolecular.
Erickson and Kahn [Citation8] first calculated the activation energies of HSC(S)OEt with MP2 and Hartree–Fock at the 6-31G(d) level. Velez et al. [Citation9] investigated the Ei reaction of EtSC(S)OMe and its substituent effects (such as α-CN, β-CN, trans-stilbene and cis-stilbene) at different theoretical levels (HF, B3LYP, MPW1K, MP2, MP3, MP4, CCSD, CCSD(T)) with different basis sets (6-31G*, 6-31G**, 6-311G**,6-311++G**, cc-pVDZ). Wu et al. [Citation10] investigated the computational study of regioselectivity for the pyrolysis of xanthates.
However, previous studies have focused on various computational methods or different transition states, but no studies have been considered regarding the quantum mechanical studies of the kinetics and thermodynamics of gas phase thermal decomposition of ethyl dithiocarbonates through a procedure devised by Adejoro and Bamkole [Citation11] using a semi-empirical method in Mopac.
2 Computational procedure
2.1 Conformational search
A conformational search was performed on the molecule to locate the structure with the lowest energy. The conformational search was carried out using a molecular mechanics force field (MMFF), which is quite successful at assigning low energy conformers and providing quantitative estimates of conformational energy differences [Citation15]. Using ab initio methods, the geometry of the lowest conformer of the reactant was optimized. Five different conformers were obtained, as shown in .
Scheme 2 Different conformers of O-ethyl, S-methyl xanthates (dithiocarbonates).
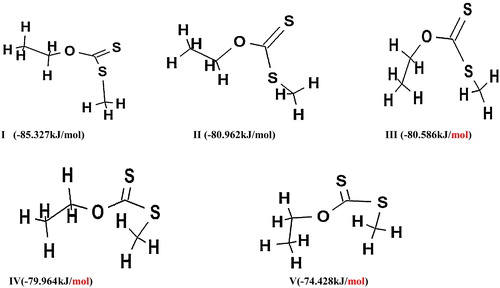
In , H1 is the β-hydrogen atom to be eliminated from the alkyl part and the dihedral is acute negative (−11.35°).
Scheme 3 Reactant, transition and product state.
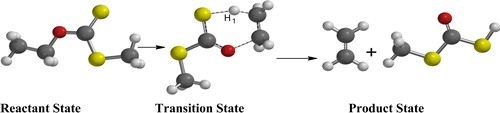
2.2 Reaction path study
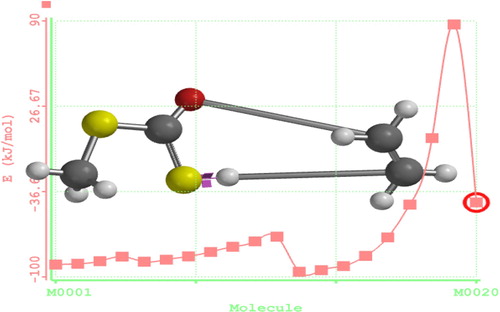
Reaction path calculations were performed on the optimized geometry of ethyl (dithiocarbonates) xanthate using H1–S10 as the reaction coordinate. The internal coordinate was varied from its initial distance in the stable reactant form to its distance in the product molecule. For instance, for ethyl xanthates, the initial distance between H1–C2 is 4.7342 Å. The inter-atomic distance is slowly altered throughout the reaction path, varying from 4.723 (initial distance) to 1.200 Å, its approximate distance in the stable product molecule, over 20 steps. Reports in previous works [Citation16] show that instead of the energy passing smoothly through a maximum, it rose to a very high value and then the geometry suddenly dropped to that of the product with a drop in the heat of formation to a value that was approximately the same as the sum of the expected products (methyl dithiocarbonates) and ethylene, as shown in .
Scheme 4 Reaction path coordinate of O-ethyl, S-methyl xanthates (dithiocarbonates).
2.3 Transition state structure
Transition states correspond to energy maxima. However, the underlying principle is that stable molecules (energy minima) will be interconnected by a smooth pathway passing through a well-defined transition state with the product. Using the guess-transition state in Spartan on the suggested mechanism of the transition state, the structure was optimized and was subjected to two tests to verify that their practical geometry corresponds to a saddle point (transition structure) and that this saddle point corresponds to the reactants and products. It is shown that the Hessian matrix of second energy derivations with respect to the coordinates yields one and only one imaginary IR frequency, which will be in the range of 400–2000 cm−1, and that the normal coordinates corresponding to the imaginary frequency smoothly connect the reactants and products. The intrinsic reaction coordinates (IRC) method was also used, which was obtained by optimizing the molecule subject to a fixed position along the reaction coordinates [Citation12]. The transition state was confirmed using the IRC method. Two IRC calculations were performed. The first calculation, a positive perturbation, evaluated the initial perturbation on the atomic coordinates in the direction of the single negative frequency, while the calculation evaluated a negative perturbation along the same normal coordinates.
2.4 Mechanism of decomposition of alkyl dithiocarbonates (xanthates)
The Chugaev reaction is analogous to the thermal decomposition of carboxylic esters of alcohols and of other related derivatives of alcohols, such as carbamates and carbonates. The mechanism of dithiocarbonate (xanthate) pyrolysis is concerted fragmentation, but products could conceivably arise from β-hydrogen abstraction to either thiol or thione sulphur atoms in mechanisms A and B, respectively, in .
In mechanism (A), the reaction occurs in one step, giving the products of reaction (1); in mechanism (B), the initial products are an alkene and unstable dithiocarbonate derivative that subsequently decompose to carbonyl sulphide and a thiol respectively. Huckel et al. [Citation2] postulated the first mechanism (A); Barton and Cram [Citation3,Citation4] proposed the second mechanism. Experimental evidence showing that a thione, rather than a thiol, sulphur atom attacks the β-hydrogen was obtained by Bader and Bourns, who performed a study of 34C and 13C isotope effects in the pyrolysis of trans-2-methyl-1-indanyl xanthate of natural isotopic abundance.
3 Calculation
Thermodynamic calculations were obtained for ethyl xanthate by calculating the ground state, GS; transition state, TS; and product. The mechanically calculated enthalpy and entropy were used, which is not a true representation of the total energy of the molecule, knowing that the major portion of enthalpy in a molecule is contained in its bonds and physical conformation; hence, the sum of the ground state energy (GSE) and mechanically calculated enthalpy are used to arrive at a closer approximation of the energy of the molecule. With the modified version of the heat of reaction, the equation is given as shown below (Spartan guide). The enthalpy of a species is defined as(1)
(1) where the superscript “sm” is the mechanically calculated enthalpy; substituting this into the initial definition of the heat reaction we have
(2)
(2) The enthalpy of reaction was calculated as 623 K.
The activation energy (Ea) was calculated according to the transition state theory for a unimolecular reaction at 623 K.(3)
(3) The entropy of the reaction was calculated by taking the difference of the product and reactant entropies, that is
and
The Gibbs free energy was calculated using the modified version of the heat of reaction equation knowing that G = H − TS.
(4)
(4) Classic transition-state theory (TST) was selected to calculate the rate constant, k(T) for the rate-determining steps, as shown in Eqs. Equation(4)
(4)
(4) and Equation(5)
(5)
(5) , which assumes that the transmission coefficient is equal to unity:
(5)
(5)
(6)
(6) where kB, h and R are the Boltzmann constant, Planck’s constant and universal gas constant, respectively, and ΔG*(T) is the standard free energy of activation at the absolute temperature T.
Derived from TST, the activation energy, Ea, and Arrhenius factor, A, could also be obtained from Eqs. Equation(3)(3)
(3) and Equation(4)
(4)
(4)
(7)
(7)
(8)
(8)
4 Results and discussion
Pyrolysis of ethyl xanthates (dithiocarbonates) gives ethane and methyl dithiocarbonates, CH3SCOSH, which subsequently decompose to give methyl sulphide and thiol. In the structure (), H1 is the hydrogen attached to the β-carbon that is hydrogen eliminated from the alkyl group. The bond between C2 and H1 is stretched with bond lengths of 1.2970 (AM1) and 1.3390 (PM3) for semi-empirical, and pyrolysis of ethyl xanthates gives methyl dithiocarbonates (CH3SCOH) and ethylene C2H4. The decomposition proceeds through a concerted fragmentation in which the product could conceivably be from β-hydrogen abstraction through a six-membered cyclic transition state that involves S–H bond formation plus C–H and C–O bond breaking, as shown in .
Fig. 1 Reaction path calculation.
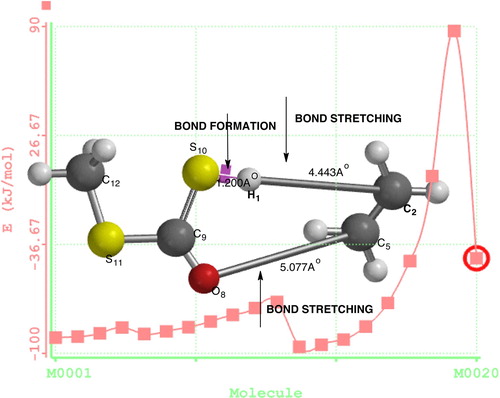
In the transition state, there is a stretch between the C2–H1 bond with bond lengths of 1.2970 (AM1), 1.3390 (PM3), (HF321G), 1.5860 (DFT/B3LYP/631G*) 1.2490 (DFT/631G**), and 1.1980 (DFT/631+G*), and against bond lengths of 1.1162 (AM1), 1.0979 (PM3), 1.108, 1.0812 (HF/321-G*), 1.0941 (DFT/631G*), 1.0930 (DFT/6-31G**), and 1.0947 (DFT/6-31+G) at the ground state. shows that both the semi-empirical and ab initio methods could effectively predict the bond length except for DFT, which has values lower compared with others from the table. Dotted lines show the point at which cleavage occurs, between C5–O8, which also has a long stretch in bond length in the transition state for all of the methods, and between C2–H1, while a new bond is formed between S10–H1. At the ground state, the bond length is in the range of 4.7–4.9; in the transition state, the bond length is in the range of 1.0–1.4 compared to a stable product bond length in the range of 1.07–1.09. The bond angles and dihedrals are shown in and .
Table 1 Selected bond lengths (Å) for the pyrolysis of O-ethyl, S-methyl xanthate (dithiocarbonate). Δd = (TS − GS).
The charges in the transition state TS () show that H1 has the largest charge development, while C5 has the smallest charge development [Citation17]. Polarization of the C5–O8 and C2–H1 bond causes an increasing positive charge on carbon atoms C5 and C9 and causes the S10 atom to become more negative (). The greater increase of the negative charge on S10 causes increased polarization of the C2–H1 bond and places considerable positive charge on the H1 atom, such that the positive charge is delocalized over the entire O8–C5–C2 frame in the TS; C9–C10–H1 is the electron demand and supply within the GS of ethyl xanthates; the vinyl carbon becomes (S10) quite highly negative, and the β-hydrogen, H1, becomes quite acidic, so that a fast proton transfer equilibrium takes place. Formation of an intermediate by an attack from the ethyl carbon on the β-hydrogen, H1, takes place as a fast step followed by the rate-determining step ().
Table 4 Selected atomic charges (Mulliken). Δq = (TS − GS) for the pyrolysis of O-ethyl-S-methyl xanthate.
Table 5 Heat of formation for the pyrolysis of O-ethyl, S-methyl xanthate (dithiocarbonates).
There is an assumption that the interaction and charge transmission of C2–C5 is not efficient so that the sizable charge on S10 is not efficiently transmitted to the electron deficient C9; hence, a double bond slowly forms between C2–C5 [Citation17].
The calculated Arrhenius parameters are as shown in and are in good agreement with the experimental values, for instance, H calculated for DFT/B3LYP/6-31G* (162.808) DFT/6-31G** (113.720) DFT/6-31+G* (115.281), and HF/3-21G*(157.817) at 623 K compares well with the experimental data. The activation energy Ea, A and rate k as calculated are in good agreement with the experimental data.
Table 6 Arrhenius parameters for O-ethyl S-methyl xanthate (dithiocarbonates).
As stated by Moyano et al. [Citation13]. Relative variation of the bond index for the transition state for every bond i involved in the reaction in Eq. Equation(9)(9)
(9)
(9)
(9) where the superscripts R, TS and P refer to the reactant, transition state and products, respectively. Additionally, %Ev is the percentage of evolution of the bond order during the calculated mechanism, as obtained from
(10)
(10) The calculated values for the percentage of evolution are listed in for O-ethyl S-methyl dithiocarbonates. The extent of the breaking of O8–C5 is 66%, while the extent of new formation of H1–S10 bonds is 45%; this implies that breaking of bonds comes before bond formation ().
Table 7 Variation of rate of reaction (S−1) with temperature (K).
Table 8 B3LYP/6-31G* calculated Wiberg bond indices, Bi, for the reactants, transition states and products: percentage of evolution through the chemical process of the bond indices at the transition states %Ev, degrees of advancement of the transition states δBav, and absolute synchronicities Sy for pathway A for the pyrolysis of O-ethyl, S-methyl dithiocarbonates.
The average relative bond indices δBav were also calculated as a measurement of the degree of advancement of the transition state along the reaction path, i.e., the early or advanced nature of the transition state, from equation(11)
(11) where n is the number of bonds involved in the reaction. For O-ethyl S-methyl dithiocarbonates, the δBav values were higher than 0.500, indicating a late character for the transition states. These observations were consistent with the energy profile in which all three intermediates were energetically higher than their corresponding reactants and the transition states were closer to the intermediates than the reactants. The synchronous or asynchronous nature of the concerted mechanism was also considered by the synchronicity, Sy, as calculated from Eq. Equation(12)
(12)
(12)
(12)
(12) where A is the asynchronicity defined from Eq. Equation(13)
(13)
(13) by Moyano et al. [Citation13].
(13)
(13)
The synchronicity values vary from zero to one, indicating completely asynchronous and synchronous, respectively.
5 Conclusion
In conclusion, it is observed that both the ab initio and DFT methods in Spartan could be used effectively to study the kinetics, mechanism and thermodynamic and vibrational spectroscopy of O-ethyl, S-methyl dithiocarbonates. The following conclusions can be drawn from the present study. The ab initio and DFT levels predict that the thermal decomposition of O-ethyl S-methyl dithiocarbonates is a concerted yet asynchronous process.
Supplementary data
Download MS Word (546.8 KB)Notes
Peer review under responsibility of Taibah University.
References
- L.Tschugaeff (Chugaev)Ber. Dtsch. Chem. Ges.32189933323335
- W.HuckelW.TappeG.LegutkeAbspaltungsreaktionen und ihr sterischer verlaufAnn. Chem.5431940191
- D.H.R.Barton464. cis-Elimination in thermal decompositionsJ. Chem. Soc.194921742178
- D.J.CramStudies in stereochemistry. IV. The Chugaev reaction in the determination of configuration of certain alcoholsJ. Am. Chem. Soc.71194938833889
- E.R.AlexanderA.MudrakStudies on the mechanism of Chugaev and acetate thermal decomposition. I. cis- and trans-2-PhenylcyclohexanolJ. Am. Chem. Soc.7219501810
- E.R.AlexanderA.MudrakStudies on the mechanism of Chugaev and acetate thermal decompositions. II. cis- and trans-2-Methyl-1-tetralolJ. Am. Chem. Soc.7219503194
- R.F.W.BaderA.N.BournsA kinetic isotope effect study of the Tschugaef reactionCan. J. Chem.391961348
- J.A.EricksonS.D.KahnTheoretical studies of thermal syn elimination reactions. The relative rates of ethyl formate, ethyl xanthate, and ethyl phosphinate eliminationsJ. Am. Chem. Soc.11619946271
- E.VelezJ.QuijanoR.NotarioJ.MurilloJ.F.RamírezComputational study of the mechanism of thermal decomposition of xanthates in the gas phase (the Chugaev reaction)J. Phys. Org. Chem.212008797
- P.WuX.ChenJ.LiY.HuangTheoretical studies on the pyrolysis of thiocarbonatesJ. Comput. Theor. Chem.103020146773
- I.A.AdejoroT.O.BamkoleSemi-empirical quantum mechanical, molecular orbital method using MOPAC, calculation of the Arrhenius parameters for the pyrolysis of some alkyl acetatesJ. Appl. Sci.5200515591563
- K.FukuiFormulation of the reaction co-ordinateJ. Phys. Chem.74197041614163
- A.MoyanoM.A.PericasE.ValentiAtheoretical study on the mechanism of the thermal and the acid-catalysed decarboxylation of 2-oxetanones (beta lactones)J. Org. Chem.541989573582
- T.LaueA.PlagensNamed Organic reaction second ed.2005john willey & son ltdChichester
- W.J.HehreGuide to Molecular Mechanics and Quantum Chemical Calculations Hardcover2003
- R.DMcIverEvidence of migration of hydrocarbons in Pleistocene sediments of theshatsky plateauvol. vi1971U.S Government printing OfficeWashington1327
- V.M.LeeM.J.CardenW.W.SchlaepferJ.O.TrojanowskiMonoclonal antibodies distinguish several differentially phosphorylated states of the two largest rat neurofilament subunits (NF-H and NF-M) and demonstrate their existence in the normal nervous system of adult ratsJ. Neurosci.7198734743488
Appendix A
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jtusci.2016.08.001.
Appendix A
Supplementary data
The following are the supplementary data to this article: