Abstract
Analysis of microbial epidemics has been revolutionized by whole-genome sequencing. We recently sequenced the genomes of 601 type emm59 Group A Streptococcus (GAS) organisms responsible for an ongoing epidemic of invasive infections in Canada and some of the United States. The epidemic has been caused by the emergence of a genetically distinct, hypervirulent clone that has genetically diversified. The ease of obtaining genomic data contrasts with the relatively difficult task of translating them into insightful epidemiological information. Here, we sequenced the genomes of 90 additional invasive Canadian emm59 GAS organisms, including 80 isolated recently in 2010–2011. We used an improved bioinformatics pipeline designed to rapidly process and analyze whole-genome data and integrate strain metadata. We discovered that emm59 GAS organisms are undergoing continued multiclonal evolutionary expansion. Previously identified geographic patterns of strain dissemination are being diluted as mixing of subclones over time and space occurs. Our integrated data analysis strategy permits prompt and accurate mapping of the dissemination of bacterial organisms in an epidemic wave, permitting rapid generation of hypotheses that inform public health and virulence studies.
Introduction
The recent unparalleled discriminatory power afforded by whole-genome sequencing has proven decisive for understanding aspects of bacterial outbreaks, including cholera,Citation1,Citation2 tuberculosis,Citation3 Listeria monocytogenes,Citation4 Escherichia coli O104Citation5,Citation6 and Group A Streptococcus (GAS also known as streptococcus pyogenes).Citation7,Citation8,Citation9 Decreasing costs, enhanced data output and improved protocols now make this technology the method of choice for some aspects of public health microbiology.Citation10,Citation11,Citation12 The ease of obtaining genome data permits the study of the evolutionary events contributing to bacterial clone emergence, strain differentiation, and epidemics at the full-chromosomal level in large samples.Citation13 Toward this end, we have used whole-genome sequencing of GAS as a model to understand the fine-structure and molecular architecture of epidemics occurring over many years.Citation7,Citation8
GAS is a human-specific pathogen that causes diseases ranging in severity from uncomplicated pharyngitis to life-threatening necrotizing fasciitis.Citation14 GAS strains are classified based on the aminoterminal sequence of the M protein, a polymorphic cell-surface adhesin and anti-phagocytic factor encoded by the emm gene.Citation15,Citation16,Citation17 Previously, we have used deep-genome sequencing data for 95 emm3 GAS strains integrated with epidemiological information to resolve the key molecular features of three successive epidemics caused by emm3 GAS in Ontario, Canada, over a period of 16 years.Citation7 This comparative pathogenomic analysis delineated the fine-structure molecular population genetics of these epidemics and defined relationships among the invasive strains responsible for the three epidemics.
More recently, using animal infection models and whole-genome sequencing of 601 emm59 GAS strains, we unambiguously demonstrated that a recently emerged, hypervirulent clone was responsible for a large epidemic of invasive GAS disease that began in 2006 in Western Canada and caused more than 500 invasive cases in 4 years. The epidemic wave extended rapidly to 11 Canadian provinces and territories and to three of the United States.Citation8,Citation18 The epidemic clone was distantly related to historic emm59 GAS strains, and also differed from emm59 GAS organisms isolated in other countries.Citation8,Citation19 Our genomic investigation precisely informed us about the population genomic landscape of the emm59 GAS epidemic, and permitted us to delineate patterns of geographic dissemination of strains in widely-dispersed areas. We also discovered that, as a population, the emm59 epidemic isolates have accumulated relatively few genetic polymorphisms over the years since sharing a common ancestor. One hypothesis to explain this finding is that over the course of the four years represented by the emm59 strain sample used in that previous study, sufficient time has not elapsed to produce considerable genomic differentiation. Extending the genomic analysis by incorporating whole-genome data for epidemic strains more recently isolated will help to differentiate between this and other hypotheses.
Despite the ability to sequence bacterial genomes rapidly, translating the whole-genome data into insightful epidemiological findings remains relatively time consuming and requires intensive logistical and computing resources.Citation13 In addition, incorporating critical strain or patient metadata into the analysis, and displaying the data in a more visual, intuitive and integrative fashion is challenging. Collectively, these problems hinder the clinical and public health responses to natural and accidental outbreaks, and deliberate release of infectious agents. Thus, methods to rapidly translate genome data into accurate, epidemiologically relevant information in a short period of time are needed. Here, we have attempted to address these limitations by use of whole-genome data from additional strains recovered from the ongoing emm59 GAS epidemic. Whole-genome data for 90 additional emm59 GAS strains isolated in Canada were generated and integrated into an improved pipeline for data processing and visualization. We show here that pertinent epidemiological information can be obtained rapidly and economically in meaningful clinical and public health situations.
Materials and methods
Bacterial strains
The strain collection includes 691 emm59 strains of different origins. 601 of these strains have been described in detail recently.Citation8 The additional 90 strains () were all isolated in Canada from patients with invasive GAS infections. One of these strains was isolated in 1993 in Ontario, and is thus far the oldest identified Canadian emm59 GAS organism. Nine strains were isolated during 2006–2009, the period of time covered by our previous report on the emm59 epidemic,Citation8 but had not been included in the previous study. The vast majority (n=80) of the newly analyzed strains were isolated in 2010 and 2011.
Table 1 Type emm59 GAS strains incorporated in this study
Whole-genome sequencing
DNA was extracted from each of the 90 emm59 GAS isolates and prepared for multiplexed (barcoded) sequencing as previously described.Citation9 Genome sequence data were obtained using an Illumina HiSeq2000 instrument according to the manufacturer’s instructions.
Bioinformatic analysis
Bioinformatic analysis was performed using a customized version of the Galaxy SuiteCitation20,Citation21,Citation22 running on the Amazon cloud. Namely, we created a pipeline composed of Tagdust,Citation23 FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) and FastX (http://hannonlab.cshl.edu/fastx_toolkit/) toolkits, and used it to parse multiplexed sequencing reads, remove barcode information and perform run quality control analyses. Polymorphism discovery was performed using Variant Ascertainment Algorithm.Citation24 Reads were aligned to the emm59 GAS strain MGAS15252 (GenBank accession number CP003116) reference genome using the Mosaik assembler (http://bioinformatics.bc.edu/marthlab/Mosaik). This strain was isolated in Canada in 2008 and its genome sequenced to closure previously; thus becoming the de facto reference genome for type emm59 GAS epidemic strains.Citation8 Unaligned reads were placed into contigs using the Velvet de novo assembler.Citation25 Contigs greater than 100 nucleotides in length were then used to search the NCBI non-redundant database using BLAST.Citation26
Phylogenetic analysis
A matrix file containing the genotype of all strains at each polymorphic locus was created from the Variant Ascertainment Algorithm polymorphism output data using a custom script. Insertions and deletions (indels) were not considered for phylogenetic analysis. Single-nucleotide polymorphisms (SNPs) were concatenated in order of occurrence relative to the reference genome and converted to a multiFASTA sequence. ClustalWCitation27 was used to align and generate a guide tree for the FASTA sequences. Neighbor-joining phylogenetic trees followed by bootstrap analysis of 1000 replicates were created using SplitsTreeCitation28 and graphically edited with TreeDyn.Citation29 The Neighbor-joining phylogenetic tree for Canadian epidemic strains isolated 2006–2011 was exported to Path-O-Gen v1.3 (http://tree.bio.ed.ac.uk/software/pathogen) and a linear regression plot for isolation date versus root-to-tip distance was generated as described by Mutreja et al.Citation30
Data visualizations
Visualizations were created using the resources offered by cBio Inc.’s Pathogen Annotated Tracking Resource Network located at http://www.patrn.net/. Illustrations were created using TIBCO Spotfire and Adobe Illustrator.
Results
Invasive infections caused by emm59 GAS remain abundant in Canada
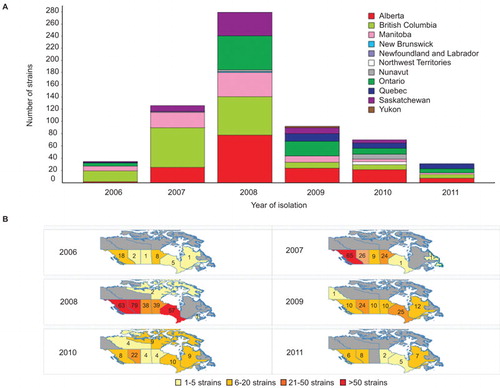
We reported previously that emm59 GAS strains were almost non-existent in Canada before 2006, but caused more than 500 invasive infections countrywide from that date until early 2010.Citation8,Citation18 The epidemic reached its peak in 2008, after which the number of cases began to decline (). However, emm59 GAS strains continued to cause invasive infections, with 56 newly identified cases in 2010 (total of 70 cases for 2010), and 32 cases in 2011. Since surveillance for GAS is passive in Canada, the actual number of emm59 GAS invasive cases is likely to have been higher in the period under investigation. During 2010 and 2011, cases occurred in vastly dispersed geographical areas covering most of the country ().
Figure 1 Number of emm59 GAS recorded cases in Canada and their geographic distribution since the beginning of the epidemic. (A) The epidemic began in year 2006 and reached its peak in 2008. By that year, cases of invasive disease were recorded over the vast majority of the country. The number of cases started to decline since 2009. However, in years 2010 and 2011, strains could still be recovered in relatively large numbers. (B) Temporospatial display of strain origin. Provinces in which emm59 GAS strains were isolated are color coded based on the number of strain isolated each year. Numbers indicate the exact number of strains isolated in a particular year in each province.
The oldest Canadian emm59 GAS strain identified is not the immediate ancestor of the epidemic emm59 clone
Our previous genome investigation of emm59 GAS strains recovered in Canada unambiguously demonstrated that regardless of geographic location or year of isolation, epidemic Canadian emm59 strains have descended from a recent, single ancestral cell that underwent rapid geographic dissemination.Citation8 The last common ancestor remains unidentified, but it was remarkable that the genomes of the epidemic strains and those of the only seven emm59 strains isolated in Canada before the epidemic began (1997–2004) were closely related.Citation8 We sequenced here the genome of an emm59 strain isolated in Canada in 1993, thus far the oldest Canadian emm59 organism identified. The genome sequence data show that this strain is genetically distantly related to the epidemic strains and thus is not the recent ancestor. Indeed, compared to reference epidemic strain MGAS15252, the 1993 emm59 GAS strain had 194 SNPs and 20 indels in the core genome (i.e., the ∼1670 kbp portion of the genome lacking mobile genetic elements that is conserved in gene content across all sequenced emm GAS typesCitation31). Phylogenetic analysis using SNP data for the core genomes found that this historic Canadian isolate also is not closely related to non-epidemic emm59 GAS organisms isolated in other geographic locations such as the United States, Japan or Spain ().
Figure 2 (A) Inferred genetic relationships among strains based on 1443 concatenated SNP loci identified by whole-genome sequencing. Newly sequenced strains all cluster within the epidemic clonal complex, with the exception of a Canadian emm59 GAS strain isolated in 1993. This strain is also genetically distant from invasive strains from Spain, Japan and the United States. Scale bar represents genetic distance. (B) Magnification of the epidemic complex part of the tree, showing the geographical area of isolation (Canadian province or US state). Strains recovered during years 2010–2011 are highlighted. In general, these more recent strains are found in the outermost part of every tree branch.

Comparative genome sequencing of new Canadian epidemic emm59 strains
The 89 additional emm59 GAS Canadian organisms differed from the reference strain, on average, by 10 SNPs and 3.4 indels (median 12 SNPs, 3 indels). The notable exception was strain MGAS24525, isolated in Manitoba in 2010, which had 465 SNPs and 71 indels relative to the reference strain. However, the vast majority of these polymorphisms mapped to mobile genetic elements, namely, prophages present in the reference sequence. Indeed, compared with the core genome of the reference strain, the number of polymorphisms was reduced to only 30 SNPs and 5 indels. Moreover, 15 of these 30 SNPs mapped to sequences in close vicinity of prophage boundaries. These results suggest that strain MGAS24525 is a bona fide emm59 GAS epidemic strain that has very recently acquired a different prophage. Phylogenetic analysis using the whole-genome data showed that the Canadian emm59 GAS organisms clustered tightly in the epidemic complex ().
Epidemic Canadian emm59 GAS strains continue to undergo clonal expansion
One of the findings of our previous work was that, as a population, the emm59 epidemic isolates have accumulated relatively few genetic polymorphisms over the years since sharing a last common ancestor. One hypothesis to explain that finding is that over the course of the 4 years represented by the emm59 strains used in the previous study, sufficient time has not elapsed to produce considerable genomic differentiation. Adding to the analysis data for 80 additional strains isolated in 2010–2011 provided us with the ability to test the hypothesis over a period of time of almost 6 years. Phylogenetic analysis of strains isolated during 2010–2011 found that these recent strains were located, in general, toward the tip of multiple branches of the phylogenetic tree, implying that they have accumulated additional SNPs compared to emm59 GAS organisms isolated previously (). When we divided the strain collection based on isolation year and calculated the average number of SNPs separating the epidemic strains isolated in a given year from the reference strain, we noticed an increase in the number of polymorphisms differentiating the epidemic strains from the reference strain over time. For example, epidemic strains recovered in 2006 differed from the reference genome, on average, by 7.6 SNPs, whereas strains isolated in 2011 differed, on average, by 13.1 SNPs. To test the hypothesis in more detail, we performed a linear regression analysis on all epidemic strains isolated since 2006 to calculate the rate of SNP accumulation on the basis of the date of isolation and the root-to-tip distance. Consistent with the hypothesis, linear regression analysis showed a steady rate of SNP accumulation (R2=0.6067; ) in the core genome.
Figure 3 Linear regression plot showing the correlation (R2) between the root-to-tip distance (y-axis) and the date of the epidemic strains (x-axis).

Use of whole-genome data to study temporospatial spread of emm59 GAS subclones
Among the critical questions that whole-genome sequencing and subsequent phylogenetic analyses can address is whether an apparent epidemic represents the emergence of a single distinct genotype, or, instead, it is caused by multiple clones that emerged simultaneously. In our initial investigation, we unambiguously demonstrated the emm59 GAS epidemic was caused by the emergence of a distinct genetic clone.Citation8 The extensive whole-genome data also permitted us to delineate clear patterns of geographic dissemination of derivative subclones.Citation8 We inferred that these geographic patterns were the result of the rapid spread of subclones in distinct geographic regions of the country. We hypothesized that the geographical patterns will begin to be obscured over time as a result of geographic mixing of subclones linked to human travel. clearly exemplifies how whole-genome data support this hypothesis: a subclone that before 2009 was composed only of strains isolated in the provinces of Alberta and British Columbia has expanded to four other Canadian provinces and territories. Mixing of the subclones is also clearly revealed among strains isolated in Quebec. Type emm59 GAS had been isolated in Quebec in 1996 and 2004, particularly in northern areas of the province. Quebec interrupted the provincial GAS surveillance program during 2005–2009. We discovered that none of the 28 emm59 GAS invasive cases reported in the province since 2009 was caused by direct lineal descendants of the strains originally identified in Quebec. Rather, they were caused by strains of genetically divergent emm59 subclones isolated first in other parts of Canada and subsequently introduced into the province on at least four different occasions (). Notably, we identified five strains whose genomes were essentially identical. Four of these strains were isolated from patients receiving care in the Montreal area, whereas the fifth was from a patient in Ontario. This is incontrovertible evidence that the strains have shared a very recent common ancestor and, also, most likely represents that the patients infected by these strains were connected to one another, either directly or via an unknown third-party donor. Temporal analysis revealed that the Ontario case was the last to be isolated (), indicating a reversal of the west to east route of transmission identified in our previous study.Citation8
Figure 4 (A) Geographic patterns of strains dissemination are diluted as the epidemic progresses over time. The highlighted areas of the tree exemplifies how specific subclones, previously restricted to specific geographical areas of Canada before 2009, are now found over vast regions of the country. Triangles represent emm59 GAS strains not considered for the analysis. (B) Epidemic strains isolated in the province of Quebec since 2009 were introduced into the province in at least five different occasions. Strains from Quebec are depicted in blue and their different locations in the phylogenetic tree highlighted by the black ellipses. With the exception of a ‘native’ population of strains isolated before 2006, all cases in the province were the result of the introduction of multiple different emm59 GAS subclones active in other parts of Canada. (C) Introduction of the different genotypes into Quebec can be precisely mapped back to the origin strain. (D) Magnification of a region of the phylogenetic depicting a subclone composed of five strains of identical genome sequence which caused disease in the Montreal area, but was later isolated in the province of Ontario.

Convergent evolution of polymorphisms in virulence genes
The radial structure of the epidemic phylogenetic tree () suggested that most of the SNPs identified in our strain collection arose once and were transmitted vertically by descent. Polymorphisms that arise independently multiple times are of interest since they may represent genetic changes that are under evolutionary selection because they confer an advantage to the organism. We identified several examples of these polymorphisms, including a single nucleotide deletion in a homopolymeric tract of 7 Ts, predicted to result in premature termination of translation of CovS, the sensor kinase of the two-component regulatory system CovRS. This component regulatory system influences the expression of a large regulon that includes many virulence genes.Citation32 CovR acts primarily as a transcriptional repressor of virulence genes. Inactivation of covR or covS has been reported to increase GAS virulence in strains of several different emm types.Citation32 The distribution of this deletion-mutation strongly suggests that it evolved independently on least six different occasions in emm59 GAS strains (). We also identified multiple different non-synonymous SNPs in the mga gene which encodes a stand alone global regulator of GAS virulence. Mga is important at several steps of the pathogenesis of the GAS infection, and is required for full GAS virulence in a skin model of infection.Citation33,Citation34 The phylogenetic distribution of one of these SNPs (resulting in a His201Arg amino-acid replacement) strongly suggests that it arose independently at least six times ().
Figure 5 Different polymorphisms arose multiple, independent times. Polymorphism analysis identified that several strains in different branches of the phylogenetic tree present either a nucleotide deletion (del T83) resulting in premature termination of the translation of the global regulator covS (indicated in red) and/or a SNP (A601C) resulting in a nonsynonymous amino acid replacement (His201Arg) in the stand alone regulator Mga (indicated in light blue). These polymorphisms could not have been inherited by descent. Their multiple, independent acquisition within the host suggests that they confer a selective advantage.

Discussion
Our study supports the idea that performing whole-genome sequencing and data interpretation rapidly can have a major impact on the study and management of epidemics. New and improved sequencing systems permit genome sequence data to be generated economically for hundreds of strains in as little as a week. A time frame as short as 1.5 days has been reported for obtaining high-quality genome data for a small number of strains.Citation35 However, data analysis continues to be a key bottleneck in genome sequence-based investigations.
During an epidemic, irrespective of whether it is natural or caused by the accidental or deliberate release of a pathogenic organism, there is substantial pressure to obtain high-quality genome and linked epidemiologic information very rapidly. It is therefore important to identify exactly what is being traced at as high resolution as possible. Important questions to be addressed are: Is this a clonal outbreak or are several pathogen genetic backgrounds involved? What is the nature and extent of organism evolution during the course of the epidemic? Here, using an improved data analysis pipeline, we have been able to accurately identify polymorphisms for 90 strains in less than 24 h after obtaining the raw data from the sequencing instrument. As importantly, we were able to integrate new and preexisting data along with geographical and temporal strain metadata to rapidly generate phylogenetic trees and other visualizations that translated the data into intuitive and insightful epidemiological information. By applying this analysis, we were able to differentiate among emm59 GAS subclones causing disease in different and geographically dispersed parts of Canada, and obtain new information about how these clones are expanding and mixing over time.
Two other important issues when studying an epidemic are identifying the origin of the organism(s) causing it and the ability to exclude or confirm genetic modifications. By limiting our analysis to the province of Quebec, we demonstrated that whole-genome sequencing, phylogenetic and temporospatial analysis were able to assign the different cases reported in the province to at least five different subclones of emm59 GAS. Moreover, we unambiguously demonstrated transmission of a particular subclone which has been active for months in the Montreal area to the province of Ontario. This type of crucial information is important for providing input on public health maneuvers, for understanding strain spreading and for inferring whether an epidemic is naturally caused or potentially the result of deliberate release of a pathogenic organism.
Whole-genome sequencing of large numbers of strains causing epidemics has also proven useful to characterize many biological properties of the organisms under investigation and is revolutionizing both virulence factor discovery and characterization and our understanding of bacterial pathogenesis.Citation36,Citation37,Citation38 In our analysis of the emm59 GAS epidemic in Canada, we rapidly identified several polymorphisms that arose multiple, independent times in the strains studied. Among others, we discovered such polymorphisms in global regulators of GAS virulence, such as a nucleotide deletion in the covS gene and a SNP resulting in a non-synonymous amino-acid change at position 201 of the predicted translated sequence of the standalone regulator Mga. Selection within the host of these polymorphisms may denote that they are of importance for the pathogenesis of infection. Although evaluating the potential involvement of these naturally occurring polymorphisms in GAS virulence requires further time and effort, it is worth noting that whole-genome sequencing analysis can lead to the quick generation of research hypothesis bearing on pathogenesis even during the initial analysis of epidemics.
In this study, we limited our analysis to genome data and temporospatial isolation information for bacterial strains. It would be possible, however, to rapidly build a well-curated database integrating other metadata such as, for example, disease manifestation and patient information, including patient genome data. Such efforts are currently underway in some clinical settings.Citation13 Whole-genome sequencing and integrative metadata analysis holds the promise of monitoring the spread and evolution of pathogens in real time, facilitating management of disease and patient treatment and continuing to provide leads for future research aimed at deciphering the virulence arsenal of pathogenic bacteria. The strategy we used here is a step forward in the route toward automation of whole-genome sequencing data analysis and provides a framework upon which to build more intuitive, rapid and precise analytic tools.
Acknowledgments
We thank the following hospitals or provincial public health laboratories for contributing GAS isolates to this study: British Columbia Centre for Disease Control, Vancouver, British Columbia, Canada; Saskatchewan Disease Control Laboratory, Regina, Saskatchewan, Canada; Cadham Provincial Laboratory, Winnipeg, Manitoba, Canada; Public Health Ontario Laboratories, Toronto, Ontario, Canada; Laboratoire de santé publique du Québec, Ste-Anne-de-Bellevue, Quebec, Canada; Stanton Territorial Hospital Laboratory, Yellowknife, Northwest Territories, Canada. We thank Concepcion C. Cantu from the Methodist Hospital Research Institute for technical help. This work was supported by The Methodist Hospital. Nahuel Fittipaldi is funded in part by a Postdoctoral Fellowship granted by the Canadian Institutes of Health Research, Ottawa, Ontario, Canada.
Notes
Abbreviations: Lt, left; Rt, right.
- Dasgupta A, Banerjee R, Das S, Basak S.Evolutionary perspective on the origin of Haitian cholera outbreak strain. J Biomol Struct Dyn2012;30:338–346.
- Chin CS, Sorenson J, Harris JBet al.The origin of the Haitian cholera outbreak strain. N Engl J Med2011;364:33–42.
- Gardy JL, Johnston JC, Ho Sui SJet al.Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med2011;364:730–739.
- Gilmour MW, Graham M, van Domselaar Get al.High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics2010;11:120.
- Rasko DA, Webster DR, Sahl JWet al.Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med2011;365:709–717.
- Rohde H, Qin J, Cui Yet al.Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N Engl J Med2011;365:718–724.
- Beres SB, Carroll RK, Shea PRet al.Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci USA2010;107:4371–4376.
- Fittipaldi N, Beres SB, Olsen RJet al.Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol2012;180:1522–1534.
- Shea PR, Beres SB, Flores ARet al.Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci USA2011;108:5039–5044.
- Loman NJ, Constantinidou C, Chan JZet al.High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol2012;10:599–606.
- Otto TD.Real-time sequencing. Nat Rev Microbiol2011;9:633.
- Baldry S.Attack of the clones. Nat Rev Microbiol2010;8:390.
- Olsen RJ, Long SW, Musser JM.Bacterial genomics in infectious disease and the clinical pathology laboratory. Arch Pathol Lab Med2012;136:1414–1422.
- Olsen RJ, Shelburne SA, Musser JM.Molecular mechanisms underlying group A streptococcal pathogenesis. Cell Microbiol2009;11:1–12.
- Lancefield RC.The antigenic complex of Streptococcus haemolyticus : I. Demonstration of a type-specific substance in extracts of Streptococcus haemolyticus. J Exp Med1928;47:91–103.
- Scott JR, Pulliam WM, Hollingshead SK, Fischetti VA.Relationship of M protein genes in group A streptococci. Proc Natl Acad Sci USA1985;82:1822–1826.
- Manjula BN, Acharya AS, Fairwell T, Fischetti VA.Antigenic domains of the streptococcal Pep M5 protein. Localization of epitopes crossreactive with type 6 M protein and identification of a hypervariable region of the M molecule. J Exp Med1986;163:129–138.
- Tyrrell GJ, Lovgren M, St Jean Tet al.Epidemic of group A Streptococcus M/emm59 causing invasive disease in Canada. Clin Infect Dis2010;51:1290–1297.
- Fittipaldi N, Olsen RJ, Beres SB, van Beneden C, Musser JM.Genomic analysis of emm59 group A Streptococcus invasive strains, United States. Emerg Infect Dis2012;18:650–652.
- Blankenberg D, von Kuster G, Coraor Net al.Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol2010; Chapter 19: Unit 19.10. 1–21.
- Giardine B, Riemer C, Hardison RCet al.Galaxy: a platform for interactive large-scale genome analysis. Genome Res2005;15:1451–1455.
- Goecks J, Nekrutenko A, Taylor J.Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol2010;11:R86.
- Lassmann T, Hayashizaki Y, Daub CO.TagDust—a program to eliminate artifacts from next generation sequencing data. Bioinformatics2009;25:2839–2840.
- Nusbaum C, Ohsumi TK, Gomez Jet al.Sensitive, specific polymorphism discovery in bacteria using massively parallel sequencing. Nat Methods2009;6:67–69.
- Zerbino DR, Birney E.Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res2008;18:821–829.
- Altschul SF, Madden TL, Schaffer AAet al.Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res1997;25:3389–3402.
- Thompson JD, Higgins DG, Gibson TJ.CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res1994;22:4673–4680.
- Huson DH, Bryant D.Application of phylogenetic networks in evolutionary studies. Mol Biol Evol2006;23:254–267.
- Chevenet F, Brun C, Banuls AL, Jacq B, Christen R.TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics2006;7:439.
- Mutreja A, Kim DW, Thomson NRet al.Evidence for several waves of global transmission in the seventh cholera pandemic. Nature2011;477:462–465.
- Beres SB, Musser JM.Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS ONE2007;2:e800.
- Churchward G.The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol Microbiol2007;64:34–41.
- Luo F, Lizano S, Banik S, Zhang H, Bessen DE.Role of Mga in group A streptococcal infection at the skin epithelium. Microb Pathog2008;45:217–224.
- Hondorp ER, McIver KS.The Mga virulence regulon: infection where the grass is greener. Mol Microbiol2007;66:1056–1065.
- Koser CU, Holden MT, Ellington MJet al.Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med2012;366:2267–2275.
- Georgiades K.Genomics of epidemic pathogens. Clin Microbiol Infect2012;18:213–217.
- Dettman JR, Rodrigue N, Melnyk AH, Wong A, Bailey SF, Kassen R.Evolutionary insight from whole-genome sequencing of experimentally evolved microbes. Mol Ecol2012;21:2058–2077.
- Musser JM, Shelburne SA3rd.A decade of molecular pathogenomic analysis of group A Streptococcus. J Clin Invest2009;119:2455–2463.