
Abstract
Of the seven known species of human retroviruses only one, human T-cell lymphotropic virus type 4 (HTLV-4), lacks a known animal reservoir. We report the largest screening for simian T-cell lymphotropic virus (STLV-4) to date in a wide range of captive and wild non-human primate (NHP) species from Cameroon. Among the 681 wild and 426 captive NHPs examined, we detected STLV-4 infection only among gorillas by using HTLV-4-specific quantitative polymerase chain reaction. The large number of samples analyzed, the diversity of NHP species examined, the geographic distribution of infected animals relative to the known HTLV-4 case, as well as detailed phylogenetic analyses on partial and full genomes, indicate that STLV-4 is endemic to gorillas, and that rather than being an ancient virus among humans, HTLV-4 emerged from a gorilla reservoir, likely through the hunting and butchering of wild gorillas. Our findings shed further light on the importance of gorillas as keystone reservoirs for the evolution and emergence of human infectious diseases and provide a clear course for preventing HTLV-4 emergence through management of human contact with wild gorillas, the development of improved assays for HTLV-4/STLV-4 detection and the ongoing monitoring of STLV-4 among gorillas and for HTLV-4 zoonosis among individuals exposed to gorilla populations.
Introduction
Human infection with a plethora of simian retroviruses is well documented and has led to global pandemics, exemplified by the human immunodeficiency virus (HIV) and human T-lymphotropic virus (HTLV), and local outbreaks, as occurs with simian foamy virus (SFV), simian T-lymphotropic virus (STLV) and occasionally simian immunodeficiency virus (SIV).Citation1,Citation2,Citation3 Given the demonstrated pandemic potential of retroviruses, a full understanding of the epidemiology and animal reservoirs of zoonotic primate retroviruses is of importance for monitoring and preventing future retrovirus pandemics.
The primate origin of these zoonotic infections has been identified by detailed epidemiological and phylogenetic analyses of both human and non-human primate (NHP) retroviruses. Through detailed examination of SIV infections in over 45 NHP species, it has been demonstrated that HIV-1 arose from multiple cross-species transmissions of SIVcpz and SIVgor from chimpanzees and gorillas, to people in west Central Africa.Citation1,Citation2 Similarly, HIV-2 in West Africa originated from multiple introductions of related SIVs from sooty mangabeys.Citation1,Citation2 Many studies have described ongoing zoonotic infection of primate workers, hunters and butchers with SFV from a variety of monkeys and apes and phylogenetic analysis showing that the co-evolution of SFV with NHPs has facilitated accurate identification of the simian origin of infection.Citation3,Citation4,Citation5,Citation6,Citation7,Citation8,Citation9,Citation10 Likewise, phylogenetic analysis has demonstrated that at least three of the four major HTLV lineages, HTLV-1, HTLV-2 and HTLV-3, originated from multiple introductions of their simian virus counterparts in monkeys and apes, STLV-1, STLV-2, and STLV-3, respectively.Citation3,Citation6,Citation7,Citation8 Thus, STLV and HTLV lineages are called primate T-lymphotropic viruses (PTLVs).
Of the four major HTLV lineages, the majority of human infections are caused by HTLV-1, which is conservatively estimated to be responsible for 5–10 million global human infections.Citation11 HTLV-2, while less frequent, has also spread globally and like HTLV-1, is transmitted from mother to child through breast feeding, sexual contact and contaminated blood products during transfusion or injection drug use.Citation11 HTLV-1 causes adult T-cell leukemia, HTLV-associated myelopathy/tropical spastic paraperesis and other inflammatory diseases in 2%–5% of infected persons.Citation12,Citation13 HTLV-2 is less pathogenic, but is increasingly associated with HTLV-associated myelopathy/tropical spastic paraperesis and some inflammatory diseases.Citation14 HTLV-3 is much less frequent and has only been found in people in Cameroon who have direct contact with NHPs from which they were likely infected.Citation6,Citation7,Citation8,Citation15,Citation16 No direct evidence of disease or secondary transmission of HTLV-3 has been reported.Citation3,Citation6,Citation7,Citation8
The fourth HTLV lineage, HTLV-4, was discovered in 2005 in a 48-year-old male hunter (1863LE) from rural Cameroon who reported hunting monkeys, chimpanzees, gorillas and other animals.Citation15 The infection of individual 1863LE currently represents the only known human or simian virus in this lineage. As for HTLV-3, person-to-person transmission and disease have yet to be reported for HTLV-4, though both viruses possess genes and motifs believed to be associated with disease potential in HTLV-1 and HTLV-2.Citation8,Citation16,Citation17,Citation18 Molecular dating suggests that HTLV-4 is the oldest PTLV lineage, having originated some 200 000 years ago (ya).Citation16 The ancient age of the HTLV-4 lineage led to the hypothesis that HTLV-4 is a descendent of an ancestral PTLV that infected humans during their evolutionary history and frequent NHP contact in Africa and represents a rare strain circulating within people in the central African region.Citation16 Alternatively, it has been hypothesized that HTLV-4 originated from a more recent zoonotic infection with a divergent STLV also present in NHPs cohabitating the forests of Cameroon.Citation16 Identification of the simian origin of HTLV-4 would help determine the validity of each hypothesis.
While STLV has been found in at least 30 African and Asian NHPs, discovery of the simian counterpart of the recently identified divergent HTLV-4 lineage found in a hunter from Cameroon has been elusive, despite surveys of potential monkey reservoirs.Citation19,Citation20,Citation21 In an attempt to discover how HTLV-4 emerged, and to shed light on its evolution and pandemic potential, we screened a large number of monkey and ape blood specimens from Cameroon using a recently developed HTLV-4-specific quantitative polymerase chain reaction (qPCR) assay.Citation22
MATERIALS AND METHODS
Ethics statement
Specimen collection from captive and wild primates was approved by the University of Johns Hopkins Animal Care and Use Committee (FS03M221 and FS06H205), University of California Institutional Animal Care and Use Committees (IACUC) (ARC#2007-110-01A, ARC#2007-110-2, ARC#2007-110-3), the University of California Davis IACUC (Protocol #16067) and the Cameroon government. All sample collections from captive primates were undertaken by veterinary animal care staff (authors Babila Tafon and John Kiyang) in the Limbe Wildlife Centre and Mfou National Park (wildlife rescue centers) during immobilization as part of quarantine health checks, regular health checks or during transport between enclosures.
Specimens from hunted primates were obtained by opportunistic sampling of animals in the wildlife trade or animals killed by local hunters as part of an ongoing effort to gather samples from bushmeat for detection of viruses to help elucidate the risks of this activity. Generally, these samples were self-collected by the hunter and gathered by members of our team at a later time point. Hunters never received money for specimens and particular care was taken to fully avoid promoting these activities. Our team had absolutely no control over the species selected by hunters, and also worked closely in the field with staff from the Cameroonian ministry responsible for wildlife to ensure that it was clear to hunters which species were protected and should not be hunted. Nonetheless, a small number of protected species were hunted in these sites; these samples were obtained and tested along with other samples. We work with the communities in Cameroon and regionally in Central Africa to address such issues and actively discourage hunting of protected species in the sites where we work. All protocols were developed in partnership with and with guidance from leading conservation organizations, including the Wildlife Conservation Society, the University of California Davis and EcoHealth Alliance (formerly, the Wildlife Trust) to ensure the highest ethical standards as we conduct trainings and build wildlife surveillance capacity.
Sample collection and preparation
Specimen collection from captive and wild animals was approved by IACUC and the Cameroon government. People who reported hunting or butchering wild animals in rural areas in Cameroon volunteered to collect blood samples from wild animals that they had hunted.Citation20 Hunters were educated about the risks associated with direct contact with wild animals and about appropriate prevention measures. Dried blood spots were collected on Whatman paper during butchering and allowed to air dry before storage in labeled envelopes. Preliminary identification of hunted species was undertaken by using pictures of animals common in the region and lists of local names of various hunted species were compiled.Citation21 Host species identity was confirmed for positive specimens using analysis of mitochondrial sequences (detailed below). Additional blood samples were obtained by technicians directly from hunted animals when encountered in the rural villages, as well as from captive wild animals orphaned by the bushmeat trade and housed at two sanctuaries in Cameroon. Samples from captive animals were obtained during routine veterinary procedures such as quarantine health checks, recaptures and during illness. Whole blood samples obtained by technicians from captive animals were collected in EDTA tubes and processed into plasma and buffy coat within 48 h and frozen at ≤−20 °C. All dried blood spot samples were stored in clip lock plastic bags with silica gel at room temperature since collection. Nucleic acids were extracted manually using the Qiagen QIAamp DNA minikit or with the Invitrogen Dynabead SILANE viral nucleic acid kit according to manufacturer's protocols. DNA integrity was confirmed by β-actin PCR as described elsewhere.Citation21 All animal samples were stored in laboratory spaces where only animal specimens were kept. All DNA preparation was conducted in spaces dedicated to animal samples. DNA concentrations were measured using a Nanodrop spectrophotometer.
PCR detection and generation of complete viral genomes
In total, 5 μL of each extracted DNA (∼100 ng) specimen was first screened using a specific qPCR assay created to detect and quantify HTLV-4 polymerase (pol) sequences in a specimen.Citation22 Positive samples were confirmed using nested PCR assays employing primers based on the HTLV-4 genome (GenBank accession number EF488483);Citation15,Citation16 primers 1863PF5 (5′-ACC ACT ACC CTT GCC TCG CCC TCC-3′) and 1863PR1 (5′-AAT GGG GAT GGT GGG GGT GGT TTC-3′) and 1863PF1 (5′-TCC AGA TTC CCC TCC CAA AGC GAT-3′) and 1863PR2 (5′-ACG ATG ACG GAT GTG GGG AAG GCT-3′) amplify 488-bp and 195-bp pol sequences, respectively. In addition, tax sequences were detected using the nested PCR primers 1863TF1 (5′-CTC CTT CTT TCA GTC CGT GCG GAG-3′) and 1863TR1 (5′-GGG GTA GTC AGG TTT GGC TGG TAT-3′) and the internal primers 1863TF2 (5′-CCT ACC GCA ACG GAT GTC TTG AAA-3′) and 1863TR2 (5′-TAT GGC GCC GGT GTG ATG ATA AAG-3′) to generate 399-bp and 275-bp fragments, respectively. Partial long terminal repeat (LTR) sequences 524-bp in size were amplified using the nested PCR primers 1863LF1 (5′-GCG ACA GCA AGC CCC CAA GGA CAA-3′) and 1863LR1 (5′-GGA GGG GTT TGA GTA CAG CGG GCT-3′) in the first round and 1863LF4 (5′-ACC TCG CCC TTA CCC ACT TCC CCT-3′) and 1863R1 in the second round of PCR. Nested PCR was also used to generate 319-bp envelope (env) sequences using generic PTLV primers FGENVF2 and PGENVR2 described previously.Citation17 Standard PCR conditions were used for 35 cycles in each round for all PCR assays. Complete STLV-4 genomes were obtained using high fidelity Taq polymerases and long PCR with primers and conditions previously used to obtain the full-length genome of HTLV-4.Citation16
NHP species confirmation, gorilla mitochondrial haplotype determination and sex determination
NHP identity was confirmed for positive individuals by amplification and sequencing of the mitochondrial DNA (mtDNA) cytochrome oxidase subunit II gene that was previously shown to be an excellent marker for primate phylogeny.Citation4 Gorilla population affinity was determined by phylogenetic analysis of mtDNA hypervariable region 1 (HV1) sequences and comparison to documented population variability.Citation23 New HV1 sequences and those available from gorillas at GenBank were aligned with ClustalW in MEGA5, edited manually, and the best model of nucleotide substitution determined to be Tamura-Nei and gamma distributed rates. Neighbor-joining trees were inferred using MEGA5 with tree topology support determined using 1000 bootstrap replicates. Nuclear mtDNA HV1 sequences were determined using this initial analysis and were then excluded from the final analysis. Sex of the wild gorillas was determined using a combination of three PCR primers in a single reaction (UTXY (forward), 5′-TGC TAC CTC AGG TGG ACA ACA AGG-3′, UTY (reverse), 5′-TGC TTG TTT CAG GCA CCA AGG RTC TAT K-3′ and UTX (reverse), 5′-CTC GAC ACT GGC AGT GCT GTT AGG-3′) to amplify the ubiquitously transcribed tetratricopeptide repeat protein gene as described in detail elsewhere.Citation24 The assay detects tetratricopeptide repeat protein sequences in humans and NHPs and produces two fragments of 127-bp and 86-bp in size for males and a single 127-bp sequence for females.
PTLV phylogenies
Nucleotide alignments were made using ClustalW in the alignment editor program in MEGA5 and edited manually. Percent nucleotide identities were calculated using alignments of selected sequences using the software Geneious Pro v6.0.3. The best fitting distance model of nucleotide substitution for each alignment was inferred using the maximum likelihood method with goodness of fit measured by the Bayesian information criterion in MEGA5. The best fitting nucleotide substitution model for the phylogenetic alignments was inferred to be the Hasegawa–Kishino–Yano model with discrete gamma and invariant among-site rate variation. Phylogenetic relationships were inferred using Bayesian analysis with the BEAST v1.6.2Citation25 program. Statistical support for the inferred Bayesian trees was assessed by posterior probabilities. For the Bayesian analyses, a relaxed or strict molecular clock model was used and each run consisted of two independent 100×106 Markov chain Monte Carlo generations with sampling every 10 000 generations and a constant or Yule coalescent tree prior. Convergence of the Markov chain Monte Carlo was assessed by calculating the effective sampling size of the runs using the program Tracer v1.5 (http://beast.bio.ed.ac.uk/Tracer). All parameter estimates showed significant effective sampling sizes >200. The tree with the maximum product of the posterior clade probabilities (maximum clade credibility tree) was chosen from the posterior distribution of 10 001 sampled trees after burning in the first 1000 sampled trees with the program TreeAnnotator version 1.6.2. The most recent common ancestor dates (TMRCAs) and nucleotide substitution rates were co-inferred using BEAST and a strong prior of the divergence time of 40 000–60 000 ya for the Melanesian HTLV-1 lineage (HTLV-1mel) and 15 000–30 000 ya for the most recent common ancestor of HTLV-2a/HTLV-2b native American strains as previously described.Citation16,Citation17,Citation26,Citation27,Citation28 Such divergence dates were based on well-established genetic and archaeological evidence, suggesting that ancestors of indigenous Melanesians and Australians migrated from Southeast Asia or the introduction of ancestral indigenous Indians into North America via the Bering Strait during those times.Citation16,Citation17,Citation26,Citation27,Citation28 The use of two calibration points has previously been shown to provide more reliable estimates of PTLV substitution rates and divergence dates than a single calibration date.Citation16,Citation18,Citation26,Citation27,Citation28 Comparisons of the tree likelihoods for each tree prior and clock model were performed using Bayes factors calculated in BEAST.
Serology
Cross-reactive HTLV antibodies were detected in plasma samples available from PCR-positive captive gorillas using the HTLV Blot 2.4 (MP Diagnostics, Santa Ana, CA, USA) Western blot kit following the manufacturer's instructions. The HTLV Blot 2.4 kit contains as antigens an HTLV-1 crude lysate, a recombinant envelope protein (Env, GD21) that most PTLVs cross react with and type-specific peptides that differentiate between HTLV-1 and HTLV-2. This kit has been used to determine the Western blot reactivity of plasma from the HTLV-4-infected person 1863LECitation15,Citation30 and plasma from HTLV-4(1863LE) was also run for comparison.
RESULTS
Prevalence and geographic range of STLV-4 infection in wild and captive gorillas from Cameroon
To broadly screen simians for potential STLV-4 infection, samples from a total of 1107 individual NHPs (681 wild and 426 captive) consisting of 21 species were screened for evidence of PTLV-4 infection using a qPCR assay specific for the detection of HTLV-4-like pol sequences ().Citation22 The pol qPCR assay has been previously shown to have a dynamic linear range of nine logs and can reliably detect at least 10 copies of HTLV-4 in 1 ug DNA.Citation22 Of the 21 species tested, DNA samples from only three wild (09584, 09948 and 19200) and three captive (Benito, Nyum and Shai) gorillas were positive using this assay (). Thus, the prevalences of STLV-4 in captive and wild gorillas in our study population were 8.3% and 13.6%, respectively. BLAST analysis of mtDNA cytochrome oxidase subunit II sequences confirmed the gorilla (Gorilla gorilla, ggo) origin of all six PTLV-4-positive animals. All positive wild gorillas were found to be males using sex-specific PCR, while the sex of the three positive captive gorillas was confirmed using the same assay with only Nyum being female ().
Figure shows the sampling sites for the three wild STLV-4-positive gorillas, the origin of the three captive gorillas and the sampling localities of the STLV-4-negative NHPs. All six STLV-4-positive animals originated in East Cameroon in proximity to the village of the HTLV-4-infected individual. The positive samples from hunted gorillas originated in villages 12 km, 26 km and 55 km from the village of the HTLV-4-infected individual outside Lomie. The exact origins of the captive animals are unknown as they may have been moved from the area where they were originally captured. Nyum was confiscated from an illegal ape dealer in Belabo (235 km north of person 1863LE), Shai was confiscated from a hunter in Abong Mbang (150 km north of person 1863LE) and Benito came from a forestry camp near Mindourou (90 km northeast of person 1863LE) (Figure ). Analysis of mtDNA HV1 sequences showed that the origin of all six gorillas were within the range of haplotypes previously observed in Cameroon and whose diversity is restricted by major rivers within Cameroon (Figure ).Citation23,Citation29 For example, Nyum is haplotype C1 which occurs north of the Dja and Sanaga Rivers; Shai, Benito and wild gorilla 19200 are all haplotype C2 which has been shown to exist between the upper Dja River and the Sanaga River, while wild gorillas 9584 and 9948 are haplotype D2 which occurs south of the Sanaga River in Cameroon and near the Cameroon/Central African Republic/Republic of Congo border (Figure ).Citation23,Citation29 These results are consistent with the presumed origin of the six positive gorillas and also verify the specimens are from six different individuals.
Figure 1 Location of STLV-4-positive gorillas and STLV-4-negative non-human primates in Cameroon. Mitochondrial hypervariable region 1 haplotypes are included following animal names or specimen numbers (i.e. C1, C2 and D2). Triangles show location where captive animals were confiscated which may not be the same location the animals were caught in the wild.
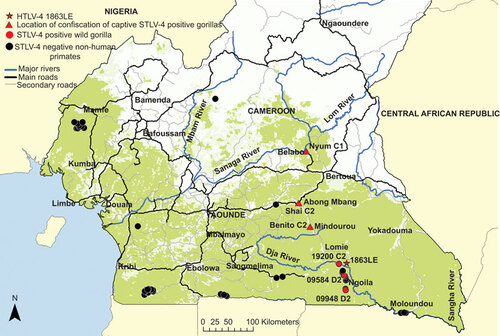
Figure 2 Gorilla mitochondrial hypervariable region 1 haplotype determination. Haplotypes inferred by neighbor-joining analysis using an alignment of eight new sequences from the current study and 158 reference sequences available at GenBank. Final alignment length was 210-bp. Bootstrap support ≥70 is provided at branch nodes. Nuclear mtDNA sequences were not included in the analysis. Gorilla sequences 51461 and 51323 are from different time points from the same animal (Nyum), whereas the two sequences from gorilla 9948 are different clones prepared at different times to verify the robustness of the cloning procedure and phylogenetic analysis.
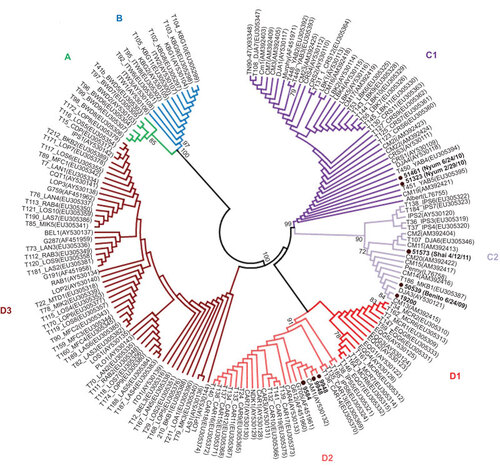
Table 1 Prevalence of STLV-4 in NHPs from Cameroon
Epidemiological and clinical considerations of STLV-4 in captive gorillas
Nyum and Shai reside in the same enclosure in the same sanctuary near Yaounde, while Benito is in a sanctuary in Limbe and has had no known contact with the other two animals, or any individuals from their group. Shai arrived at the sanctuary in 2002; a blood sample from 2009 was negative by qPCR testing and subsequent samples at three separate time points in 2011 were qPCR-positive (). Nyum arrived in April 2003 and has shared the same enclosure with Shai since September 2003. Nyum tested qPCR-positive in 2009. No earlier samples are available for either of these animals. Nyum's first observed sexual encounter was in June 2012; however, no activity with Shai has been observed. Nonetheless, these results suggest that Shai may have infected Nyum in captivity. Benito arrived in 1996 and a single blood sample from 2009 was qPCR-positive. Blood specimens from 32 additional wild-born and one captive-born captive gorillas, including all wild-born animals that share enclosures with the positive animals at the two sanctuaries, and including the animals that arrived at the same time as positive individuals, were tested and were all found to be negative for STLV-4 sequences.
Figure 3 HTLV Western blot serologic pattern of STLV-4-infected captive gorillas from Cameroon. Asterisks indicate specimens with indeterminate Western blot results. Positive controls are those supplied with manufacturer's kit. Reactivity to HTLV-specific proteins is indicated on the left.
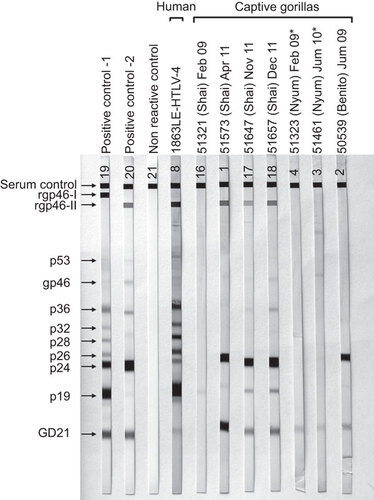
To further investigate infection in the STLV-4-positive gorillas, we tested plasma samples available from the three captive gorillas using an HTLV-1/2 Western blot kit previously shown to detect antibodies in the HTLV-4-infected hunter. We found a broad range of cross-reactivity in the STLV-4-positive samples to the HTLV-1/2 antigens (Figure and ). Samples from Shai and Benito were seropositive showing reactivity to the p24 Gag and GD21 Env proteins (Figure ). The 2009 sample from Shai was weakly seroreactive to the p19 Gag protein only, whereas his subsequent samples in 2011 were all HTLV-2-like with reactivity to the HTLV-2-type specific peptide (rgp46-II) reflective of possible seroconversion near the first time point in 2009 with infection likely obtained from Nyum in captivity. Plasma from Benito showed a seropositive but untypeable pattern without reactivity to the rgp46 type-specific peptides. Both plasma samples from Nyum were seroindeterminate and were weakly seroreactive to only the GD21 recombinant Env protein. Plasma from HTLV-4 (1863LE) was also HTLV-2-like as previously reported,Citation14,Citation30 but in contrast to the STLV-4-infected gorillas, also showed reactivity to the p26–p32 Gag proteins.
We also determined proviral loads (pVLs) in the STLV-4-infected gorillas and HTLV-4-infected hunter using our qPCR test to evaluate the potential role of increased pVLs in transmission and disease progression as occurs with other retroviruses, like HTLV and HIV. pVLs detectable in the gorillas ranged from 56 copies/100 cells to 7348 copies/100 cells (mean=3647 copies/100 cells; median=3849 copies/100 cells) (). pVLs in wild-caught gorilla 9584 were below the limit of detection. In contrast, pVLs in the HTLV-4-infected hunter (1863LE) were very low (0.295 copies/100 cells) compared to the gorilla pVLs. The two gorillas, Nyum and Shai, which shared the same enclosure had comparable pVLs averaged from their multiple timepoints (4453 and 3229 copies/100 cells, respectively), but a trend towards increased pVLs over time was observed in both animals.
The potential for PTLV-4 to cause disease like HTLV-1 and HTLV-2 is not clear. The only HTLV-4-infected person was apparently healthy in 2006; however, his family reported in 2009 that he had passed away after a short illness, characterized by fatigue and respiratory symptoms. Animal records showed that all three captive gorillas with STLV-4 infection experienced illnesses common in wild and captive animals, including wounds from aggressive behavior, skin infections, colds, fever and conjunctivitis. However, Nyum has developed some lameness and possible brain damage after an acute illness with fatigue, fever, seizures, lethargy, deafness, loss of vision and temporary paralysis in 2005. It is unclear if the high pVLs in this animal are associated with these possible neurological symptoms. Blood samples were not available from 2005 for qPCR testing when these symptoms were reported.
Table 2 Identification of PTLV-4 in gorillas from Cameroon
Phylogenetic analysis of STLV-4 and dating of the evolutionary history of PTLV-4
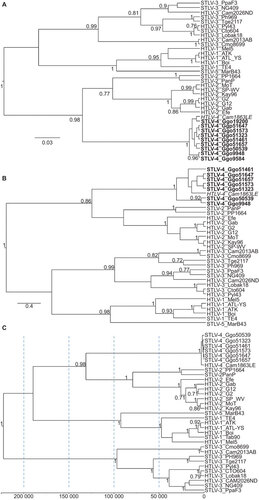
Partial PTLV-4 pol, env and tax sequences were detected by nested PCR for the seven gorilla samples () and all showed at least 99% nucleotide identity to HTLV-4 (1863LE) and to each other with the exception of gorilla 9584 which had a PTLV-1-like env sequence (see below). PTLV-4 LTR sequences from all six animals, which are typically one of the most divergent regions in PTLV since it is a non-coding region and is not under the same evolutionary constraints as coding regions, shared 98.4%–99.5% nucleotide identity with the HTLV-4 LTR. The greatest LTR sequence divergence was seen within the wild gorillas and ranged within 97.9%–99.5% compared to 99.8%–100% in the captive gorillas. Combined, these results confirm the identification of STLV-4 and demonstrate limited diversity of STLV-4 in different gorilla populations in Cameroon. These results were confirmed by phylogenetic analysis of tax and pol sequences which showed a clustering of all STLV-4 sequences with those from HTLV-4 (Figure ).
Figure 4 Identification of STLV-4 in gorillas from Cameroon. Phylogenetic trees inferred using Bayesian analysis in the BEAST program and a relaxed molecular clock. (A) Topology inferred using a 225-bp alignment of tax sequences; (B) topology inferred using a 376-bp alignment of first and second coding position pol sequences; (C) topology and divergence dates inferred using first and second codon positions of concatenated gag, pol and env sequences. Scale bars show units of time and for the concatenated sequences is years to the most recent common ancestors backwards in time from PTLV sequences. Posterior probabilities ≥0.7 are provided at branch nodes.
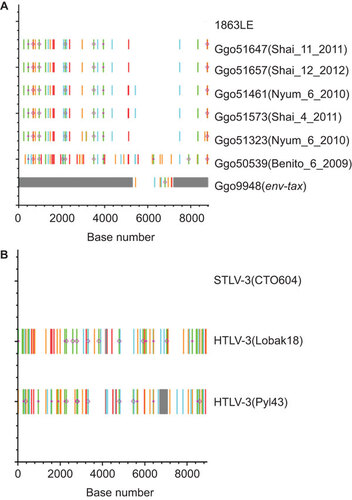
To further demonstrate the genetic relatedness of STLV-4 and HTLV-4 and to investigate changes that may occur following cross-species transmission, we obtained complete genomes from all three captive animals (Benito, Nyum and Shai), including complete genomes from Nyum and Shai at multiple time points allowing an investigation of longitudinal virus evolution (). All complete STLV-4 genomes were 8793–8794 bp in length, were nearly identical to each other and shared 99.4%–99.7% nucleotide identity to the 8791-bp genome of HTLV-4 (1863LE) due to 28–50 point mutations throughout the genome, but not affecting the open reading frames. 1919-bp of the env-tax region was obtained from one wild gorilla (9948), which showed at least 99.3%–99.5% nucleotide identity to STLV-4 from the captive gorillas and 99.5% identity to HTLV-4 (1863LE). Five of the nucleotide substitutions in the complete genomes were G>A mutations likely introduced via ABOPEC deamination, but which did not result in the introduction of premature stop codons (Figure ). Twice as many G>A mutations were also present in the sequences of two hunters found to be infected with STLV-3 most likely from a red-capped mangabey (Figure ). The HTLV-3 genome of one these individuals (Pyl43) also contained a 366-bp deletion in the pX proximal region that may also be further evidence of host restriction, but sequencing of STLV-3 genomes from additional mangabeys is needed to verify this hypothesis. Nonetheless, these results suggest the absence of gross genetic mutations that would affect infectivity and replication of PTLV-4.
Figure 5 Detection of nucleotide substitutions and APOBEC-induced hypermutation in alignments of complete and partial PTLV genomes. Analysis performed using the Hypermut and Highlighter programs at the HIV sequence database at the Los Alamos website (http://www.hiv.lanl.gov/). Sequence differences and evidence of hypermutation were determined by comparison with the complete genome of (A) HTLV-4(Cam1863LE) or (B) STLV-3(CTO604) and are highlighted as follows: A, Green; T, red; G, orange; C, light blue; IUPAC, dark blue; gaps, gray; circle, APOBEC; diamond G>A. Base number is shown on x-axis, y-axis has no units and is used for only locating reference and query sequences.
To estimate the possible date of STLV-4 entry into humans, detailed Bayesian phylogenetic analyses were performed. Sequence analysis showed unequal base composition for some lineages and substitution saturation at the third codon position (cdp) for all PTLVs similar to that observed by others.Citation16,Citation17 However, substitution saturation was not observed in the first and second cdps and these sites were thus suitable for estimating posterior evolutionary rates and divergence dates of PTLV by using Bayesian analysis with an Markov chain Monte Carlo algorithm. Phylogenetic analysis of concatenated gag–pol–env first and second cdp sequences from PTLV-4 and prototypical PTLVs showed strong clustering of STLV-4 with HTLV-4 (1863LE) and separate lineages for PTLV-1, PTLV-2 and PTLV-3 (Figure ) consistent with analysis of the shorter gene regions. To estimate the possible date of introduction of STLV-4 into humans, we inferred divergence dates using a Bayesian framework and four combinations of clock models and tree priors. Bayes factor analysis showed that the relaxed clock model with a Yule process speciation model fit the data better for the TMRCA estimates than the relaxed clock model with a constant tree prior and both molecular clock models (2ln BF=97.23). Using these methods, we found that the PTLV posterior mean evolutionary rates ranged from 4.68×10−7 to 13.13×10−7 substitutions/site/year (), which are consistent with previous estimates.Citation8,Citation16,Citation17 Divergence of the progenitor of the PTLV-4 and PTLV-2 lineages using the relaxed clock model and a Yule tree prior occurred about 131 000 ya and were somewhat higher using the three other models (), but were within the TMRCA date ranges previously determined by our group using analyses of separate gene regions.Citation16 In contrast, TMRCA estimates for the PTLV-4 ancestor are relatively more recent occurring about 3300–8400 ya, suggesting a relatively recent introduction of STLV-4 into humans ().
Table 3 PTLV evolutionary time-scale inferred using Bayesian analysis and first and second codon positions of concatenated gag, pol and env genes
STLV-1 and STLV-4 co-infection in one wild gorilla
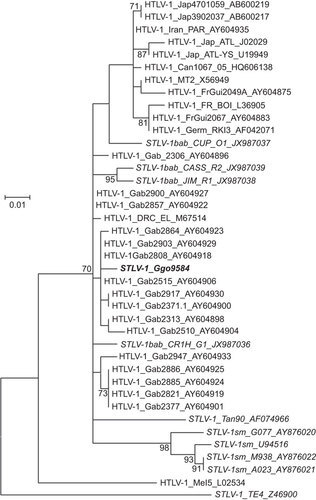
Sequence analysis of the partial env fragment (326-bp) obtained using PTLV generic PCR primers from gorilla 9584 showed that this animal was infected with both STLV-1 and STLV-4. The env sequences of this gorilla shared 98% nucleotide identity to STLV-1 from olive baboons (Papio anubis), which have a broad geographic distribution, including Cameroon. The 9584Ggo sequence also showed 99% nucleotide identity to HTLV-1 subtype F env sequences from Gabon,Citation31 which was confirmed by phylogenetic analysis (Figure ). We consistently obtained STLV-1 env sequences in repeat PCR reactions using dried blood spot DNA from this gorilla. However, examination of all sequences obtained from this animal showed no evidence of heterogeneity as would be expected in a mixed infection, suggesting that all primers used for testing samples from this gorilla may have partial selective specificity to either STLV-1 or STLV-4, which may also explain why we were unable to amplify all STLV-4 sequences from this animal using HTLV-4-specific primers.
Figure 6 STLV-1 co-infection of a wild gorilla from Cameroon. Identification of STLV-1 genotype in a wild gorilla using maximum likelihood phylogenetic analysis of 325-bp envelope sequences. Reference sequences were available at GenBank and accession numbers are provided in sequence names. One thousand non-parametric bootstrap replicates were used to assess the strength of the branching order. Bootstrap values ≥70 are provided at branch nodes. Scale bar is substitutions per site. PTLV-1 subtypes are provided. Country codes are: Gab, Gabon; Jap, Japan; FrGui, French Guinea; FR, France; Can, Canada; Germ, Germany; Mel, Melanesia. Simian codes are: sm, sooty mangabey; bab, baboon; Ggo, gorilla; Tan, tantalus monkey.
Sequence accession numbers
All new STLV-4 and G. gorilla mtDNA sequences generated in the current study have been deposited at GenBank with the accession numbers (KF029402–KF029433).
DISCUSSION
Prior to this report, HTLV-4 was the only known human retrovirus without a recognized animal reservoir. Previous efforts to identify HTLV-4-like viruses in a variety of African NHPsCitation19,Citation20,Citation21 may have been unsuccessful because they relied on HTLV-1-based serological assays and generic PTLV PCR assays with presumably limited sensitivities for STLV-4 detection. In addition, previous studies were limited in that they largely examined monkeys rather than gorillas or other apes for PTLV infection, since blood specimens are not as readily available from ape species.Citation19,Citation20,Citation21 The predominance of ape specimens from previous simian retrovirus studies are non-invasively collected feces and urine samplesCitation2,Citation32,Citation33 and while SIV and SFV have been successfully detected in these body fluids, the detection of PTLV in these samples has not yet been reported.Citation2,Citation32,Citation33
A combination of screening data, geographical distribution, behavioral data and phylogenetic analyses provides strong evidence that gorillas are the natural host of STLV-4 and the reservoir for the known human case of HTLV-4. Through the largest NHP screening to date for evidence of PTLV-4, we detected STLV-4 in three wild and three captive gorillas, and found no evidence of PTLV-4 from a large series of other samples from a wide range of NHP species. While our knowledge of the exact geographical origins of the three captive infected gorillas remains imperfect, when combined with the distribution of the infected wild gorillas, the data demonstrate that the individual infected with HTLV-4 lived in an endemic region of gorilla STLV-4. Furthermore, the HTLV-4-infected individual reported contact with gorillas through hunting, a behavior reported by only 11% of male respondents.Citation34 Given his self-reported behavior and the detection of PTLV-4 in gorillas from the area where the HTLV-4-infected individual lived and hunted, it is likely that his infection was the result of direct contact with gorillas through hunting. Phylogenetic analyses, including results from complete STLV-4 genomes from three gorillas, show that the gorilla STLV-4 sequences are nearly identical to that of the HTLV-4 case, lending further support to the conclusions that STLV-4 is endemic in gorillas, and that the HTLV-4 infection represents the cross-species transmission of STLV-4 from gorilla to human. While the possibility that STLV-4 in gorillas resulted from the cross-species transmission from a third species cannot be ruled out, it is unlikely given the range of NHP species screened in Cameroon.
Our results put to rest the debate over ancient origins of HTLV-4 in humans. While the PTLV-4 lineage is older than the other HTLV lineages, the data indicate that human infection resulted from a relatively recent cross-species transmission of a gorilla virus, rather than an ancient infection in human ancestors. Our findings support the importance of gorillas in the emergence and evolution of retroviruses and other human infections. In addition to HTLV-4, contact with gorillas is now thought to be associated with the origin of human infections with HIV-1 group PCitation2,Citation35 and with sporadic human infections with STLV-1ggoCitation15 and SFVgor.Citation3,Citation7,Citation9,Citation35,Citation36 Interestingly, all variants of these novel retroviruses emerged in Cameroon where a long history of contact with NHPs and hunting of gorillas exists. Furthermore, recent work on the origins of malignant malaria indicated that gorilla Plasmodium was the source of human Plasmodium falciparum.Citation37 It now appears evident that gorillas should be considered keystone reservoirs for human disease emergence along with other taxa such as chimpanzees and some groups of bats. Whether this is because of their phylogenetic proximity to humans or other elements of their population genetics, ecology or behavior remains to be determined.
Our results also have conservation and clinical implications for gorillas, an increasingly rare close relative of humans. Gorillas are endangered and protected by law throughout their ranges, and the opportunities for synergetic messages from health, conservation and law enforcement authorities should be encouraged to ensure the maximum benefit from any communication is obtained and that messages are harmonious. Considering that people continue to hunt apes in central Africa, greater efforts are needed to educate hunters on the risks of zoonotic infections to prevent the emergence of new microbes. The role that gorillas play in the emergence of human infectious disease provides an additional justification for prioritizing efforts to minimize hunting through strong enforcement of wildlife hunting regulations and work to develop appropriate and accessible animal proteins in regions where gorillas are endemic.
While the clinical outcomes of STLV-4 infection in gorillas remain preliminary, the findings suggest that STLV-4 may be transmitted between gorillas in captivity and show that STLV-4 may lead to chronic infections in gorillas. While the significance remains unclear for now, the evidence of paralysis and neurological symptoms in Nyum are intriguing because of the known potential for similar symptoms associated with HTLV-1 and HTLV-2 in humans. Nonetheless, it is now clear that SIV can lead to serious disease in chimpanzees following chronic infection,Citation2 and leukemia and lymphoma have been reported in baboons infected with STLV-1,Citation38,Citation39 so the potential for STLV-4 to also cause disease cannot be excluded. The high pVLs detected in some gorillas, including Nyum, may help explain the ease of transmissibility of STLV-4 and the disease potential in these animals. However, the two captive gorillas with high pVLs do not appear to have any clinical evidence of leukemia or lymphoma or other HTLV-1-like disease symptoms. It is unclear if the high pVLs in gorilla Nyum are associated with the possible neurological symptoms reported in this animal and will require testing of additional animals. In contrast, pVLs in the HTLV-4-infected hunter were comparatively much lower than those in the gorillas. It will be important to test close contacts of this hunter to determine if these low pVLs are associated with person-to-person transmission of HTLV-4.
Our finding of a broad range of Western blot profiles in the STLV-4-infected gorillas using HTLV-1/2 antigens suggests that these current serological assays are likely missing or misclassifying PTLV-4 infections in people and NHPs. For example, the HTLV-4 Western blot profile and those for the majority of the STLV-4-infected gorilla specimens from Shai were HTLV-2-like, suggesting that people with this Western blot profile, without follow-up PCR and sequence analysis, could be infected with PTLV-4 and not HTLV-2. While adequate specimen volumes were not available to evaluate detection of STLV-4 using available HTLV-1/2 EIAs, these Western blot results indicate that better serological assays are clearly needed to address this deficiency. However, the sensitivity and specificity of the HTLV-1/2 Western blot assay for detection of PTLV-4 infection is currently unknown.Citation30 The use of HTLV-4-specific screening PCR assays may have limited the detection of PTLV-4-related infection in some animals whose STLV-4 sequence is divergent in the qPCR primer regions. The development and use of the highly degenerate PTLV qPCR assays combined with the development of PTLV-4-specific, or improved serologic assays for broader PTLV detection, will all aid in better understanding the geographic distribution of PTLV-4 infection in humans and gorillas. Combined with our finding of a wide distribution of STLV-4 in gorillas across the forests of Cameroon, the absence of HTLV screening in Cameroon and the use of non-HTLV-4 antigens for serologic detection, suggests a potentially broader human exposure to STLV-4 in Cameroon and that additional HTLV-4-infected humans may yet be identified.
We also identified dual STLV-1 and STLV-4 infection in one wild gorilla (Ggo9584). Others have reported dual STLV-1 and STLV-3 infected monkeys in Cameroon,Citation40 suggesting that although rare, multiple infections can occur in this region of high viral and primate diversity. The significance of these co-infections is currently unknown, but could lead to genetic recombination as occurs in other dual retroviral infections. The STLV-1 in this gorilla clustered with HTLV-1 subtype F from persons living in GabonCitation41 and thus, could be the primate origin of these human infections since the geographic range of G. gorilla extends west from the Congo River into Cameroon, Gabon, Central African Republic and mainland Equatorial Guinea. Gorilla 9584 was from near the southern border of Cameroon, a border which Cameroon also shares with Gabon and primate bushmeat hunting and consumption are known to occur in Gabon, further supporting this possibility. Thus, expanded testing for STLV-4 and HTLV-4 in Gabon and other African countries where gorillas exist may be warranted.
The recent discovery of two new retrovirus species in humans (HTLV-3 and -4) and the ongoing detection of new cases of cross-species transmission of retroviruses from various animal hosts,Citation3,Citation7,Citation8,Citation9,Citation10,Citation36 indicates the regularity with which humans are acquiring novel retrovirus diversity. Given the devastating nature of the global HIV pandemic and the prior emergence of two species of HTLV that have spread globally (HTLV-1 and -2), the importance of monitoring for novel retrovirus outbreaks in human populations is paramount. Determining the animal reservoirs of zoonotic retroviruses, monitoring cross-species transmission of these viruses and others, and following individuals with primary infections all remain primary tools for the identification of new viruses with pandemic potential. Work to monitor individuals exposed to gorillas through hunting and other activities, appropriate efforts to decrease such activities and ongoing laboratory studies to further characterize STLV-4 provide a clear path towards preventing HTLV-4 emergence as a human disease.
Funding for this study was provided by the Faucett Family and Rawji Foundations and Global Viral Forecasting (ANRS12182/12255 and NIH RO1 AI50529), graciously supported by the US Department of Defense Armed Forces Health Surveillance Center, Division of Global Emerging Infections, Surveillance Operations (AFHSC GEIS), the Defense Threat Reduction Agency Cooperative Biological Engagement Program (DTRA-CBEP), Department of Defense HIV/AIDS Prevention Program (DHAPP), Google.org, the Skoll Foundation and the US Agency for International Development (USAID), whose support was made possible by the generous support of the American people through the USAID Emerging Pandemic Threats PREDICT program. Use of trade names is for identification only and does not imply endorsement by the US Department of Health and Human Services, the Public Health Service or the Centers for Disease Control and Prevention. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention or the US Government.
- Sharp PM, Hahn BH.Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med2011;1: a006841.
- Peeters M, Delaporte E.Simian retroviruses in African apes. Clin Microbiol Infect2012;18: 514–520.
- Wolfe ND, Switzer WM, Carr JK et al.Naturally acquired simian retrovirus infections in central African hunters. Lancet2004;363: 932–937.
- Switzer WM, Salemi M, Shanmugam V et al.Ancient co-speciation of simian foamy viruses and primates. Nature2005;434: 376–80.
- Switzer WM, Tang S, Ahuka-Mundeke S et al.Novel simian foamy virus infections from multiple monkey species in women from the Democratic Republic of Congo. Retrovirology2012;9: 100.
- Calattini S, Chevalier SA, Duprez R et al.Discovery of a new human T-cell lymphotropic virus (HTLV-3) in Central Africa. Retrovirology2005;2: 30.
- Calattini S, Betsem E, Bassot S et al.Multiple retroviral infection by HTLV type 1, 2, 3 and simian foamy virus in a family of Pygmies from Cameroon. Virology2011;410: 48–55.
- Zheng H, Wolfe ND, Sintasath DM et al.Emergence of a novel and highly divergent HTLV-3 in a primate hunter in Cameroon. Virology2010;401: 137–145.
- Betsem E, Rua R, Tortevoye P, Froment A, Gessain A.Frequent and recent human acquisition of simian foamy viruses through apes' bites in central Africa. PLoS Pathog2011;7: e1002306.
- Mouinga-Ondémé A, Caron M, Nkoghé D et al.Cross-species transmission of simian foamy virus to humans in rural Gabon, Central Africa. J Virol2012;86: 1255–1260.
- Gessain A, Cassar O.Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol2012;3: 388.
- Yamashita M, Ido E, Miura T, Hayami M.Molecular epidemiology of HTLV-1. J. Acquired Immune Defic Syndr Hum Retrovirol1996;13: S124–S131.
- Gessain A, Mahieux R.Epidemiology, origin and genetic diversity of HTLV-1 retrovirus and STLV-1 simian affiliated retrovirus. Bull Soc Pathol Exot2000;93: 163–171.
- Araujo A, Hall WW.Human T-lymphotropic virus type II and neurological disease. Ann Neurol2004;56: 10–19.
- Wolfe ND, Heneine W, Carr JK et al.Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc Natl Acad Sci USA2005;102: 7994–7999.
- Switzer WM, Salemi M, Qari SH et al.Ancient, independent evolution and distinct molecular features of the novel human T-lymphotropic virus type 4. Retrovirology2009;6: 9.
- Switzer WM, Qari SH, Wolfe ND, Burke DS, Folks TM, Heneine W.Ancient origin and molecular features of the novel human T-lymphotropic virus type 3 revealed by complete genome analysis. J Virol2006;80: 7427–7438.
- Calattini S, Chevalier SA, Duprez R et al.Human T-cell lymphotropic virus type 3: complete nucleotide sequence and characterization of the human tax3 protein. J Virol2006;80: 9876–9888.
- Liégeois F, Boué V, Mouacha F et al.New STLV-3 strains and a divergent SIVmus strain identified in non-human primate bushmeat in Gabon. Retrovirology2012;9: 28.
- Ahuka-Mundeke S, Mbala-Kingebeni P, Liegeois F et al.Identification and molecular characterization of new simian T cell lymphotropic viruses in nonhuman primate bushmeat from the Democratic Republic of Congo. AIDS Res Hum Retroviruses2012;28: 628–635.
- Sintasath DM, Wolfe ND, LeBreton M et al.Simian T-lymphotropic virus diversity among nonhuman primates, Cameroon. Emerg Infect Dis2009;15: 175–184.
- Duong YT, Jia H, Lust JA et al.Absence of evidence of HTLV-3 and HTLV-4 in patients with large granular lymphocyte (LGL) leukemia. AIDS Res Hum Retroviruses2008;24: 1503–1505.
- Anthony NM, Johnson-Bawe M, Jeffery K et al.The role of Pleistocene refugia and rivers in shaping gorilla genetic diversity in central Africa. Proc Natl Acad Sci USA2007;104: 20432–20436.
- Villesen P, Fredsted T.Fast and non-invasive PCR sexing of primates: apes, Old World monkeys, New World monkeys and Strepsirrhines. BMC Ecol2006;6: 8.
- Drummond AJ, Rambaut A.BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol2007;7: 214.
- Salemi M, Desmyter J, Vandamme AM.Tempo and mode of human and simian T-lymphotropic virus (HTLV/STLV) evolution revealed by analyses of full-genome sequences. Mol Biol Evol2000;17: 374–386.
- van Dooren S, Meertens L, Lemey P, Gessain A, Vandamme AM.Full-genome analysis of a highly divergent simian T-cell lymphotropic virus type 1 strain in Macaca arctoides. J Gen Virol2005;86: 1953–1959.
- Lemey P, van Dooren S, Vandamme AM.Evolutionary dynamics of human retroviruses investigated through full-genome scanning. Mol Biol Evol2005;22: 942–951.
- Clifford SL, Anthony NM, Bawe-Johnson M et al.Mitochondrial DNA phylogeography of western lowland gorillas (Gorilla gorilla gorilla). Mol Ecol2004;13: 1551–1565, 1567.
- Switzer WM, Hewlett I, Aaron L, Wolfe ND, Burke DS, Heneine W.Serologic testing for human T-lymphotropic virus-3 and -4. Transfusion2006;46: 1647–1648.
- Capdepont S, Londos-Gagliardi D, Joubert M et al.New insights in HTLV-I phylogeny by sequencing and analyzing the entire envelope gene. AIDS Res Hum Retroviruses2005;21: 28–42.
- Etienne L, Locatelli S, Ayouba A et al.Noninvasive follow-up of simian immunodeficiency virus infection in wild-living nonhabituated western lowland gorillas in Cameroon. J Virol2012;86: 9760–9772.
- Neel C, Etienne L, Li Y et al.Molecular epidemiology of simian immunodeficiency virus infection in wild-living gorillas. J Virol2010;84: 1464–1476.
- Wolfe ND, Prosser TA, Carr JK et al.Exposure to nonhuman primates in rural Cameroon. Emerg Infect Dis2004;10: 2094–2099.
- Plantier JC, Leoz M, Dickerson JE et al.A new human immunodeficiency virus derived from gorillas. Nat Med2009;15: 871–872.
- Calattini S, Betsem EB, Froment A et al.Simian foamy virus transmission from apes to humans, rural Cameroon. Emerg Infect Dis2007;13: 1314–1320.
- Liu W, Li Y, Learn GH et al.Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature2010;467: 420–425.
- McCarthy TJ, Kennedy JL, Blakeslee JR, Bennett BT.Spontaneous malignant lymphoma and leukemia in a simian T-lymphotropic virus type 1 (STLV-1) antibody positive baboon. Lab Anim Sci1990;40: 79–81.
- Voevodin A, Samilchuk E, Schätzl H, Boeri E, Franchini G.Interspecies transmission of macaque simian T-cell leukemia/lymphoma virus type 1 in baboons resulted in an outbreak of malignant lymphoma. J Virol1996;70: 1633–1639.
- Courgnaud V, van Dooren S, Liegeois F et al.Simian T-cell leukemia virus (STLV) infection in wild primate populations in Cameroon: evidence for dual STLV type 1 and type 3 infection in agile mangabeys (Cercocebus agilis). J Virol2004;78: 4700–4709.
- Capdepont S, Londos-Gagliardi D, Joubert M et al.New insights in HTLV-I phylogeny by sequencing and analyzing the entire envelope gene. AIDS Res Hum Retroviruses2005;21: 28–42.