
Abstract
Little is known regarding the molecular epidemiology of Middle East respiratory syndrome coronavirus (MERS-CoV) circulating in dromedaries outside Saudi Arabia. To address this knowledge gap, we sequenced 10 complete genomes of MERS-CoVs isolated from 2 live and 8 dead dromedaries from different regions in the United Arab Emirates (UAE). Phylogenetic analysis revealed one novel clade A strain, the first detected in the UAE, and nine clade B strains. Strain D998/15 had a distinct phylogenetic position within clade A, being more closely related to the dromedary isolate NRCE-HKU205 from Egypt than to the human isolates EMC/2012 and Jordan-N3/2012. A comparison of predicted protein sequences also demonstrated the existence of two clade A lineages with unique amino acid substitutions, A1 (EMC/2012 and Jordan-N3/2012) and A2 (D998/15 and NRCE-HKU205), circulating in humans and camels, respectively. The nine clade B isolates belong to three distinct lineages: B1, B3 and B5. Two B3 strains, D1271/15 and D1189.1/15, showed evidence of recombination between lineages B4 and B5 in ORF1ab. Molecular clock analysis dated the time of the most recent common ancestor (tMRCA) of clade A to March 2011 and that of clade B to November 2011. Our data support a polyphyletic origin of MERS-CoV in dromedaries and the co-circulation of diverse MERS-CoVs including recombinant strains in the UAE.
INTRODUCTION
Since its first appearance in 2012, Middle East respiratory syndrome (MERS) has affected more than 1300 cases in more than 25 countries in four continents, and has an alarming fatality rate of more than 30%.Citation1 A novel lineage C betacoronavirus, MERS coronavirus (MERS-CoV), has been confirmed to be the etiological agent of MERS.Citation2, Citation3 Subsequent detection of MERS-CoV and its antibodies in dromedaries in various countries in the Middle East and North Africa has suggested that these animals are probably the reservoir for MERS-CoV.Citation4, Citation5, Citation6 In addition, both before and after the MERS epidemic, the discovery of other closely related lineage C betacoronaviruses in various bat species and hedgehogs suggests that these animals may be hosts for an ancestor of MERS-CoV.Citation7, Citation8, Citation9, Citation10 In further support of this hypothesis, the spike protein of Tylonecteris bat CoV HKU4 binds dipeptidyl peptidase 4,Citation11, Citation12 the cellular receptor for MERS-CoV.
As of August 2016, 212 genome sequences of MERS-CoV were available in GenBank. Although 91 of the sequences were obtained from MERS-CoV in dromedaries, a large proportion of them were from a recent study in Saudi Arabia.Citation13 The small number of dromedary MERS-CoV genomes obtained from other countries has hindered understanding of the epidemiology and evolutionary history of the virus in camels outside of Saudi Arabia. In our previous report on MERS-CoV epidemiology in Dubai, nine dromedary MERS-CoV strains were sequenced and found to be closely related.Citation14 Recently, we have reported another dromedary MERS-CoV strain detected in an isolated dromedary herd in the United Arab Emirates (UAE).Citation15 Complete genome sequencing and phylogenetic analysis has indicated that this MERS-CoV strain is a unique member of a cluster of closely related MERS-CoV strains obtained from patients in the Hafr-Al-Batin region of Saudi Arabia and Qatar,Citation16, Citation17 as well as those from patients in the recent Korean outbreak.Citation18 From the results of these two studies, we hypothesized that diverse MERS-CoV strains may be circulating in dromedaries of the UAE. To test this hypothesis, we performed complete genome sequencing of 10 additional strains of MERS-CoV isolated from dromedaries in different regions of the UAE. The results support a polyphyletic origin of MERS-CoV in dromedaries and the co-circulation of diverse strains from multiple sources in the same farm. A novel clade A strain, the first identified in the UAE, may belong to a separate lineage, A2, circulating in dromedaries.
MATERIALS AND METHODS
Strains and viral culture
Ten MERS-CoVs isolated from 10 respiratory samples from 10 dromedaries sent to the Central Veterinary Research Laboratory in Dubai, UAE, in 2014 and 2015 were included in this study. Isolation of MERS-CoV was performed as previously described.Citation15 Briefly, the samples were diluted 10-fold with viral transport medium and filtered. Two hundred microliters of the filtrate was inoculated into 200 μL of Minimum Essential Medium (Gibco, Grand Island, NY, USA). Four hundred microliters of the mixture was added to 24-well tissue culture plates containing Vero cells for adsorption inoculation. After 1 h of adsorption, excess inoculum was discarded, the wells were washed twice with phosphate-buffered saline, and the medium was replaced with 1 mL of Minimum Essential Medium (Gibco). The cultures were incubated at 37 °C with 5% CO2 and were inspected daily by inverted microscopy for cytopathic effect (CPE) for five days. Each of the cultures exhibited the typical CPE of detachment and rounding of cells. All cultures with a CPE were confirmed to be infected with MERS-CoV through a real-time quantitative RT-PCR assay targeting a region of the viral genome upstream of the envelope gene and isothermal amplification with a Genie instrument (Optigene Limited, Horsham, UK).
RNA extraction
Viral RNA was extracted from the cultures using a QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany). The RNA was eluted in 60 μL of Buffer AVE and was used as the template for RT-PCR.
Complete genome sequencing
The complete genomes of the 10 isolated dromedary MERS-CoV strains were sequenced as previously described.Citation14, Citation15 Briefly, the RNA extracted from the MERS-CoV strains was converted to cDNA by using a combined random priming and oligo(dT) priming strategy. The cDNA was amplified by primers designed on the basis of multiple sequence alignments of available MERS-CoV genome sequences using previously described strategies.Citation19 The 5′ ends in the viral genomes were confirmed via rapid amplification of cDNA ends (RACE) using a 5′/3′ RACE kit (Roche Diagnostics GmbH, Mannheim, Germany). Sequences were assembled and edited to produce the final sequences of the MERS-CoV genomes.
Genome analysis
Nucleotide and amino acid sequences of predicted open reading frames (ORFs) and the full genomes of 10 MERS-CoV were aligned with 85 human MERS-CoV and 87 camel MERS-CoV genomes using Multiple Alignment using Fast Fourier Transform (MAFFT). Ten MERS-CoV genome sequences from GenBank were not included in the analysis because of an incomplete 5′ genomic region or having redundant sequences of a single strain. Pairwise identity of the 182 MERS-CoV genome sequences as well as their ORFs and predicted proteins was calculated using MEGA5.
Phylogenetic analysis
Maximum-likelihood phylogenetic trees with 1000 bootstrap replicates were constructed using PhyML v3.0 (Montpellier, France) on the basis of the complete genome, ORF1a, ORF1b, and S genes of 85 human and 97 camel MERS-CoV genomes. The best-fit substitution model was selected using jModelTest and used in the maximum-likelihood analysis.
Recombination analysis
To detect possible recombination, bootscan analysis was performed by using the nucleotide alignment of the genome sequences of MERS-CoV and Simplot version 3.5.1, as previously described.Citation20, Citation21 The analysis was conducted using a sliding window of 1500 nucleotides moving in 200 nucleotide steps with genome sequences obtained in the present study as the query. Possible recombination sites suggested by the bootscan analysis were confirmed through multiple sequence alignments.
In addition to the bootscan analysis, possible recombination breakpoints were also detected using RDP, GENECONV, BOOTSCAN, MAXIMUM CHI SQUARE, CHIMAERA, SISCAN and 3Seq implemented in Recombination Detection Program Version 4 (RDP4). Automasking was used for optimal recombination detection. The RDP analysis was run without a reference and with a window size of 60, the BOOTSCAN window size was increased to 500, the MAXCHI and CHIMAERA number of variable sites per window was increased to 120, and the window size and step size for SISCAN were increased to 500 and 20, respectively. Potential recombination events detected by four or more of the seven independent recombination detection methods in the ten genomes in this study were further analyzed with phylogenetic trees constructed using sequences upstream and downstream of the potential recombination breakpoint.
Estimation of divergence times
Divergence times for the MERS-CoV strains were calculated using a Bayesian Markov chain Monte Carlo (MCMC) approach implemented in BEAST (version 1.8.0), as described previously.Citation22, Citation23 One representative strain was selected for MERS-CoV strains with close sequence similarity and obtained from the same outbreak. Analyses were performed under the SRD06 substitution models for the concatenated main coding regions of the genome (ORF1ab, S, E, M and N), with an uncorrelated lognormal molecular clock and a GMRF skyride coalescent model. The MCMC run was 1 × 108 steps long with sampling every 1000 steps. Convergence was assessed on the basis of the effective sampling size after a 10% burn-in using Tracer software, version 1.6.0.Citation22 The mean time to the most recent common ancestor (tMRCA) and the highest posterior density regions at 95% (HPDs) were calculated. The trees were summarized in a target tree by using the Tree Annotator program included in the BEAST package by choosing the tree with the maximum sum of posterior probabilities (maximum clade credibility) after a 10% burn-in.
Nucleotide sequence accession numbers
The nucleotide sequences of the ten dromedary MERS-CoV genomes sequenced in this study have been submitted to the GenBank sequence database under accession nos. KX108937–KX108946.
RESULTS
Clinical and epidemiological data
We isolated 10 MERS-CoV strains from nasal swabs of two live and eight dead dromedary calves from different regions in the UAE, including a breeding herd, which returned from its winter pasture in Saudi Arabia and calves from dromedary dairy farms in Dubai and Umm Al Quwain. The clinical and epidemiological data of the 10 dromedaries infected with MERS-CoV strains isolated and sequenced in this study are summarized in and . The median age of the dromedaries was 1 month (range: 15 days to nine months). Two of the eight dead dromedaries exhibited nasal discharge before they died.
Figure 1 Geographical distribution of dromedaries in the United Arab Emirates infected with MERS-CoV strains that were isolated in the present study. The number of dromedaries corresponds to the number of MERS-CoV isolates from the sampling location.
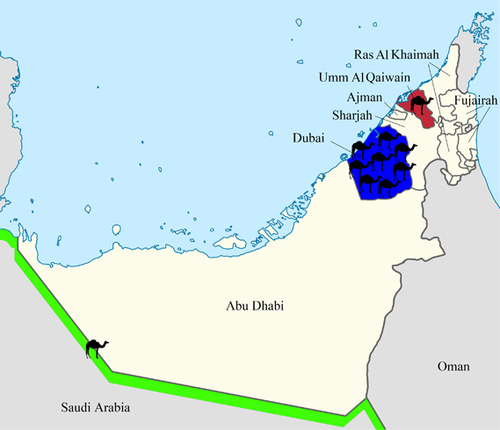
Table 1 Clinical and epidemiological data of the 10 dromedaries with MERS-CoV strains isolated and sequenced in this study
Phylogenetic analysis
Phylogenetic analysis of the complete genomes of the ten sequenced MERS-CoV isolates showed that one isolate belongs to clade A, and nine belong to clade B (; ). Phylogenetic analysis of the complete genome, ORF1a, ORF1b and the S gene sequences all showed that the clade A strain, D998/15, isolated from a 1-month-old dromedary calf from a special dairy farm in Dubai, is a unique member of clade A, being most closely related to another dromedary isolate, NRCE-HKU205, which was previously detected in Egypt (). Moreover, these two isolates formed a separate cluster distinct from the other two known clade A strains, EMC/2012 and Jordan-N3/2012, both isolated from humans. The results suggest that clade A isolates from humans and camels may form two separate lineages, A1 and A2. This hypothesis is further supported by analysis of amino acid substitutions along the whole genome sequences.
Figure 2 Maximum-likelihood phylogeny based on the complete genome sequences of 182 MERS-CoV strains. A general time-reversible model of nucleotide substitution with estimated base frequencies, the proportion of invariant sites, and the gamma distribution of rates across sites were used in the maximum-likelihood analysis. Bootstrap values are shown next to the branches. The scale bar indicates the number of nucleotide substitutions per site. MERS-CoVs from dromedaries are indicated in black circles. The ten MERS-CoV strains sequenced in the present study are colored: blue, Dubai; brown, Umm Al Quwain; green, Saudi Arabia. MERS-CoVs isolated from the United Arab Emirates are indicated by gray boxes.
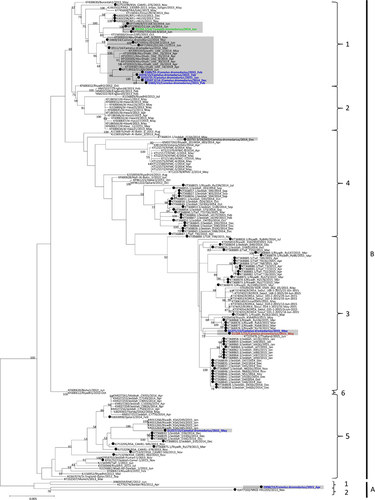
Figure 3 Maximum-likelihood phylogeny based on ORF1a, ORF1b and S gene sequences of 182 MERS-CoV strains. TIM1+I+G, GTR+I+G, and TK 2+I substitution models were selected for the ORF1a, ORF1b and S gene trees, respectively. Bootstrap values are shown next to the branches. The scale bar indicates the number of nucleotide substitutions per site. MERS-CoVs from dromedaries are indicated in black circles. The 10 MERS-CoV strains sequenced in the present study are colored: blue, Dubai; brown, Umm Al Quwain; green, Saudi Arabia. MERS-CoVs isolated from the United Arab Emirates are indicated by gray boxes.

Table 2 Comparison of amino acid substitutions between different clades and lineages
Comparison of deduced amino acid sequences of proteins among clade A1, clade A2 and the six lineages of B strains showed a total of 16 substitutions, most occurring in the viral nsp3 and S proteins (). Notably, two substitutions in nsp3, at positions 1574 and 1717, were found within the catalytic domain of PLpro.Citation24 Highly variable residues were observed at position 1574, which has been reported to be under positive selection.Citation25 Another substitution in the S protein at position 1020 (glutamine in clade A strains and histidine/arginine in clade B strains) was found within heptad repeat 1 of the S2 region. Interestingly, the heptad repeat region has recently been reported to be a major selection target among MERS-CoVs, and different residues at position 1020 may affect the stability of the six-helix bundle formed by the heptad repeats.Citation26 There were nine amino acid substitutions between clade A and B strains, and five amino acid substitutions between clade A1 and A2 strains. From these results, we propose that the two clade A dromedary isolates D998/15 and NRCE-HKU205 should be classified under a new lineage, A2, which is distinct from the two clade A human isolates EMC/2012 and Jordan/2012.
The nine clade B isolates belong to three distinct lineages (). Five isolates from a dairy farm in Dubai and one from a Dubai breeding herd returning from Saudi Arabia belong to lineage 1. One isolate from a dairy farm in Dubai and one from Umm Al Qaiwain belong to lineage 3. One isolate from a dairy farm in Dubai belongs to lineage 5 (). Phylogenetic trees based on ORF1a, ORF1b and the S gene were further constructed to assess strains that had an inconsistent phylogenetic position in the different trees. The two lineage three strains in this study (D1271/15 and D1189.1/15), along with other lineage three strains, were found to cluster more closely with lineage 4 MERS-CoV in the tree constructed for ORF1a, but cluster more closely with lineage 5 MERS-CoV in the tree constructed for ORF1b (). For the tree constructed for the S gene, these lineage three strains did not cluster closely with either lineage 4 or lineage 5 MERS-CoV (). The remaining eight strains identified in this study were found to consistently cluster within the same lineages in all three trees ().
Recombination analysis
Because phylogenetic analyses showed inconsistent phylogenetic positions for strains D1271/15 and D1189.1/15 in the ORF1a and ORF1b trees, we performed bootscan analysis and multiple sequence alignments for these two strains. For both strains, bootscan analysis showed high bootstrap support for clustering between the two strains and lineage 4 MERS-CoV in two parts of their genomes (position 1–15000); but for position 15000–24000, bootscan analysis showed high bootstrap support for clustering between the two strains and lineage 5 MERS-CoV (). A multiple sequence alignment using these two strains, a lineage 4 MERS-CoV and a lineage 5 MERS-CoV further indicated that upstream of position 14697, the two strains possessed nucleotides identical to lineage 4 MERS-CoV; but from position 16155–23785, the two strains possessed nucleotides similar to lineage 5 MERS-CoV ().
Figure 4 Detection of potential recombination events by bootscan analysis and multiple alignments. Bootscanning was conducted with Simplot version 3.5.1 (F84 model; window size, 1,500 bp; step, 200 bp) on a gapless nucleotide alignment. (A) D1271/15 and D1189.1/15 were used as the query sequences and compared with the genome sequences of a lineage 4 MERS-CoV strain Hafr-Al-Batin 1 (red, KF600628), a lineage 1 MERS-CoV strain UAE/Abu Dhabi_UAE_9 (green, KP209312) and a lineage 5 MERS-CoV strain D1157/15 (blue, KX108944). (B) D2731.3/14 was used as the query sequence and compared with the genome sequences of a lineage 1 MERS-CoV strain, FRA/UAE (green, KF745068), a lineage 4 MERS-CoV strain, KFMC-4 (red, KT121575) and a lineage 5 MERS-CoV strain, England-Qatar/2012 (blue, KC667074).
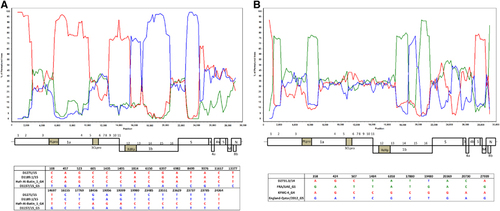
For the analysis using RDP4, recombination breakpoints were detected in the two genomes at positions 14958 and 28835. Phylogenetic trees constructed using concatenated nucleotides sequences from position 1 to 14957 and 28836 to 30087, and from position 14958 to 28835 confirmed the inconsistent phylogenetic position of these two newly detected strains in the two trees ().
Figure 5 Maximum-likelihood trees constructed using nucleotide sequences of non-recombination regions (A), and recombination regions (B) detected using RDP4. The non-recombination region is approximately positions 1 to 14957 and 28836 to 30087, and the recombination region is approximately positions 14958 to 28835, with position numbering based on the D1189.1/15 genome sequence. Lineages are indicated by colored boxes: lineage 1, green; lineage 3, red; and lineage 5, blue.

Estimation of divergence dates
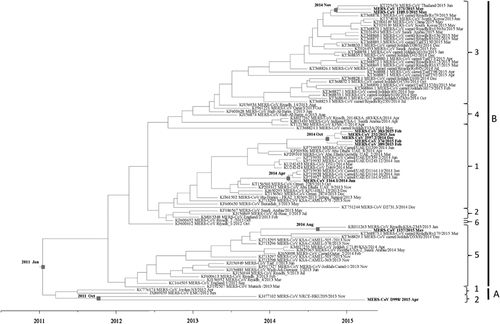
The inferred evolutionary rate of MERS-CoV was 8.55 × 10−4 (95% HPD: 7.09 × 10−4, 1.01 × 10−3). The divergence time of the present 10 dromedary strains is shown in . On the basis of time-resolved phylogeny, the root of the tree is January 2011 (95% HPD: June 2010, August 2011). The time of the most recent common ancestor (tMRCA) of clade A was dated back to March 2011 (95% HPD: August 2010, September 2011) and that of clade B to November 2011 (95% HPD: July 2011, March 2012) ().
Figure 6 Estimation of time to the most recent common ancestor for MERS-CoV. The time-scaled phylogeny was summarized from all Markov chain Monte Carlo phylogenies of the concatenated coding regions (ORF1ab, S, E, M and N) of 90 phylogenetically distinct MERS-CoV genomes, which were analyzed under the relaxed-clock model with an uncorrelated lognormal distribution in BEAST version 1.8.0. The 10 MERS-CoV genomes sequenced in this study are in boldface.
DISCUSSION
In this study, we showed that MERS-CoVs isolated from dromedaries in the UAE were of polyphyletic origin. For the eight MERS-CoV strains isolated from dromedaries from dairy farms in Dubai, one belongs to clade A (D988/15) and seven (D2597.2/14, D252/15, D374/15, D383/15, D389/15, D1157/15 and D1271/15) belong to clade B (). Interestingly, the source clade A strain, D988/15, was isolated from a young female dromedary in a special dairy farm with camel mothers from Pakistan. Therefore, the source of this virus was most probably different from that of the seven clade B strains, which were isolated from dromedaries from a different dairy farm. The latter farm is a large closed system, which may explain the relative genetic closeness of the circulating MERS-CoV strains. Yet, the seven clade B isolates belonged to three distinct lineages, with five isolates (D2597.2/14, D252/15, D374/15, D383/15 and D389/15) clustered with several other MERS-CoV from Dubai.Citation27 For the MERS-CoV isolated from a dromedary calf returning from Saudi Arabia (1164.1/14), the dromedary was from a herd of approximately 50 female breeding dromedaries in Dubai. Every year, these camels were brought to Saudi Arabia for 6 months because of the better grazing conditions due to the higher amount of rainfall in Saudi Arabia than in Dubai. The dromedary calves from this herd were born in Saudi Arabia. Young dromedaries are usually removed from the herd and start their training as racing camels when they are one year old. Although this MERS-CoV was most probably acquired in Saudi Arabia, its genome sequence is closely related to two human MERS-CoV strains from Oman (KT156560) and Abu Dhabi (KP209312), respectively (). The MERS-CoV isolated from the dromedary in Umm Al Quwain (D1189.1/15) is closely related to D1271/15 in this study and to a human MERS-CoV strain isolated in Thailand (KT225476) (). The present data showed that the genome sequences of the MERS-CoV strains isolated in the UAE were intermingled with other MERS-CoV strains found elsewhere in the Middle East or strains from patients who acquired their infections directly or indirectly in the Middle East. Moreover, diverse MERS-CoVs are co-circulating in dromedaries in the UAE, which derive from different sources.
This polyphyletic origin of MERS-CoV is unique among human CoVs. Such a polyphyletic origin of MERS-CoV in dromedaries is similar to the diverse lineages of SARS-CoV-like viruses in horseshoe bats,Citation28, Citation29 but in contrast to the monophyletic origin of most human SARS-CoV strains, MERS-CoV strains from humans are polyphyletic as a result of multiple camel-to-human transmission events.Citation30, Citation31 According to the existing evidence, a single interspecies transmission event probably occurred from bats to palm civets as the intermediate or amplification host, and then from palm civets to humans before the SARS epidemic in 2003.Citation28, Citation32, Citation33 The rapid expansion of the epidemic arose after efficient human-to-human transmission of the human SARS-CoV strains. In contrast, MERS-CoV has become endemic in dromedaries of the UAE and other countries in the Middle East, with diverse strains being introduced into the human population. This finding is in line with the overall MERS-CoV phylogeny in the Middle East, with the tMRCA of all MERS-CoVs being dated back to around January 2011, thus suggesting that MER-CoV has emerged in humans relatively recently ( and ). The difference in evolutionary pathways between SARS-CoV and MERS-CoV also provides an explanation for why, MERS—unlike SARS, which disappeared rapidly after civets were segregated from humans by closing wild animal markets in provinces in Southern China—has persisted for more than three years and will probably continue to circulate in dromedaries and humans unless effective vaccines are available. Recently, viruses closely related to human coronavirus 229E (HCoV 229E) have also been found in camels.Citation34 However, in contrast to the polyphyletic origin of human and camel MERS-CoVs, the HCoV 229E and related dromedary-derived viruses were each monophyletic, thus suggesting that this endemic human coronavirus may constitute a descendant of camelid-associated viruses.
The clade A isolate D988/15, the first clade A MERS-CoV detected in the UAE, may belong to a separate lineage, A2, within clade A. At present, there are only a few clade A isolates, including the first human MERS-CoV isolated in Saudi Arabia (EMC/2012), another human isolate from Jordan (Jordan-N3/2012), two dromedary strains from Egypt (NRCE-HKU205 and NRCE-HKU270 with partial sequence available only)Citation35 and six Nigerian strains (genome not completely sequenced).Citation36 Although clade A strains are much less common than clade B strains, our results suggest that diverse clade A MERS-CoVs may be present in various countries in the Middle East and Africa. Both phylogenetic and amino acid substitution analyses showed that the current clade A strains may consist of two distinct lineages: one from humans (A1) and one from camels (A2). Further studies to isolate and sequence more clade A strains in humans and camels may provide insights on the role of clade A strains in MERS-CoV evolution and the potential significance of the observed amino acid changes for host adaptation.
The present results show that recombination events are common among certain strains of MERS-CoV. Recombination is a well-recognized mechanism by which CoVs generate diversity. We have previously described recombination among the three human CoV HKU1 genotypes, and other human or animal coronaviruses,Citation21, Citation37, Citation38 and other researchers have also found recombination in coronaviruses such as feline coronavirus type II and infectious bronchitis virus.Citation39 A number of studies have also reported recombination among MERS-CoVs from different countries.Citation13, Citation40 However, we should be cautious when interpreting possible recombination events solely on the basis of phylogenetic analysis to avoid drawing premature conclusions. For example, by phylogenetic tree analysis, strains D1271/15 and D1189.1/15 in the present study and strain D2731.3/14, which we have reported previously,Citation15 showed inconsistent phylogenetic clustering in trees based on different viral genes (). However, bootscan analysis and multiple alignments did not reveal evidence of recombination for strain D2731.3/14 (). Only strains D1271/15 (from Dubai) and D1189.1/15 (from Umm Al Quwain) showed evidence of a likely recombination event after stepwise examination using phylogenetic analysis, bootscan analysis and multiple sequence alignments (). It is also important to note that the nucleotide sequences of all MERS-CoV genomes are >99% identical. Therefore, even for the strains with possible recombination, there are only approximately 10 base pair differences upstream and downstream of the potential recombination site between the ‘recombinant’ and ‘parent’ strains. Although these sequence differences may have arisen through recombination, they may also have resulted from individual nucleotide mutations. Further studies with careful interpretation of recombination analysis results will be important to understand the role of recombination in the emergence and evolution of MERS-CoV.
This work was partly supported by the Theme-based Research Scheme (Project NO. T11/707/15) of the University Grant Committee, by the HKSAR Health and Medical Research Fund, and by the University Development Fund and Strategic Research Theme Fund of The University of Hong Kong. We are grateful to Drs Peter Nagy and Judit Juhasz from the Emirates Industry for Camel Milk and Products (EICMP), Dubai, UAE; as well as Dr Ismail Hassabelrasoul from the Nad Al Sheba Camel Dairy Farm, Dubai, UAE; and Dr Abubaker Abbas from Umm Al Quwain, UAE.
Related Research Data
- ChanJF,LauSK,ToKKet al.Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease.Clin Microbiol Rev2015; 28:465–522.
- van BoheemenS,de GraafM,LauberCet al.Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans.mBio2012; 3:e00473–e00412.
- ZakiAM,van BoheemenS,BestebroerTMet al.Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia.N Engl J Med2012; 367:1814–1820.
- PereraRA,WangP,GomaaMRet al.Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt.Euro Surveill2013; 18:20574.
- ReuskenCB,HaagmansBL,MullerMAet al.Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study.Lancet Infect Dis2013; 13:859–866.
- WooPC,LauSK,WerneryUet al.Novel betacoronavirus in dromedaries of the Middle East, 2013.Emerg Infect Dis2014; 20:560–572.
- WooPC,WangM,LauSKet al.Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features.J Virol2007; 81:1574–1585.
- LauSK,LiKS,TsangAKet al.Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of Pipistrellus bat coronavirus HKU5 in Japanese Pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus.J Virol2013; 87:8638–8650.
- MemishZA,MishraN,OlivalKJet al.Middle East respiratory syndrome coronavirus in bats, Saudi Arabia.Emerg Infect Dis2013; 19:1819–1823.
- CormanVM,KalliesR,PhilippsHet al.Characterization of a novel betacoronavirus related to Middle East respiratory syndrome coronavirus in European hedgehogs.J Virol2014; 88:717–724.
- WangQ,QiJ,YuanYet al.Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26.Cell Host Microbe2014; 16:328–337.
- YangY,DuL,LiuCet al.Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat-to-human transmission of MERS coronavirus.Proc Natl Acad Sci USA2014; 111:12516–12521.
- SabirJS,LamTT,AhmedMMet al.Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia.Science2016; 351:81–84.
- WerneryU,CormanVM,WongEYet al.Acute Middle East respiratory syndrome coronavirus infection in livestock dromedaries, Dubai, 2014.Emerg Infect Dis2015; 21:1019–1022.
- WerneryU,RasoulIE,WongEYet al.A phylogenetically distinct Middle East respiratory syndrome coronavirus detected in a dromedary calf from a closed dairy herd in Dubai with rising seroprevalence with age.Emerg Microbes Infect2015; 4:e74.
- CottenM,WatsonSJ,KellamPet al.Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study.Lancet2013; 382:1993–2002.
- HaagmansBL,Al DhahirySH,ReuskenCBet al.Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation.Lancet Infect Dis2014; 14:140–145.
- CowlingBJ,ParkM,FangVJet al.Preliminary epidemiological assessment of MERS-CoV outbreak in South Korea, May to June 2015.Euro Surveill2015; 20:7–13.
- WooPC,LauSK,ChuCMet al.Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia.J Virol2005; 79:884–895.
- WooPC,LauSK,YipCCet al.Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1.J Virol2006; 80:7136–7145.
- LauSK,LeeP,TsangAKet al.Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination.J Virol2011; 85:11325–11337.
- DrummondAJ,RambautA.BEAST: Bayesian evolutionary analysis by sampling trees.BMC Evol Biol2007; 7:214.
- LauSK,WooPC,LiKSet al.Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports the murine origin of Betacoronavirus 1 and has implications for the ancestor of Betacoronavirus lineage A.J Virol2015; 89:3076–3092.
- KilianskiA,MielechAM,DengXet al.Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors.J Virol2013; 87:11955–11962.
- ForniD,CaglianiR,MozziAet al.Extensive positive selection drives the evolution of nonstructural proteins in lineage C betacoronaviruses.J Virol2016; 90:3627–3639.
- ForniD,FilippiG,CaglianiRet al.The heptad repeat region is a major selection target in MERS-CoV and related coronaviruses.Sci Rep2015; 5:14480.
- HunterJC,NguyenD,AdenBet al.Transmission of Middle East respiratory syndrome coronavirus infections in healthcare settings, Abu Dhabi.Emerg Infect Dis2016; 22:647–656.
- LauSK,WooPC,LiKSet al.Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats.Proc Natl Acad Sci USA2005; 102:14040–14045.
- WooPC,LauSK,LiKSet al.Molecular diversity of coronaviruses in bats.Virology2006; 351:180–187.
- AzharEI,El-KafrawySA,FarrajSAet al.Evidence for camel-to-human transmission of MERS coronavirus.N Engl J Med2014; 370:2499–2505.
- MemishZA,CottenM,MeyerBet al.Human infection with MERS coronavirus after exposure to infected camels, Saudi Arabia, 2013.Emerg Infect Dis2014; 20:1012–1015.
- GuanY,ZhengBJ,HeYQet al.Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China.Science2003; 302:276–278.
- LiW,ShiZ,YuMet al.Bats are natural reservoirs of SARS-like coronaviruses.Science2005; 310:676–679.
- CormanVM,EckerleI,MemishZAet al.Link of a ubiquitous human coronavirus to dromedary camels.Proc Natl Acad Sci USA2016; 113:9864–9869.
- ChuDK,PoonLL,GomaaMMet al.MERS coronaviruses in dromedary camels, Egypt.Emerg Infect Dis2014; 20:1049–1053.
- ChuDK,OladipoJO,PereraRAet al.Middle East respiratory syndrome coronavirus (MERS-CoV) in dromedary camels in Nigeria.2015Euro Surveill2015; 20.
- LauSK,PoonRW,WongBHet al.Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel betacoronavirus subgroup.J Virol2010; 84:11385–11394.
- LauSK,FengY,ChenHet al.Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination.J Virol2015; 89:10532–10547.
- HerreweghAA,SmeenkI,HorzinekMCet al.Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus.J Virol1998; 72:4508–4514.
- WangY,LiuD,ShiWet al.Origin and possible genetic recombination of the Middle East respiratory syndrome coronavirus from the first imported case in China: phylogenetics and coalescence analysis.mBio2015; 6:e01280–e01215.