
Abstract
Mycobacterium tuberculosis (MTB) Beijing strains have caused a great concern because of their rapid emergence and increasing prevalence in worldwide regions. Great efforts have been made to investigate the pathogenic characteristics of Beijing strains such as hypervirulence, drug resistance and favoring transmission. Phylogenetically, MTB Beijing family was divided into modern and ancient sublineages. Modern Beijing strains displayed enhanced virulence and higher prevalence when compared with ancient Beijing strains, but the genetic basis for this difference remains unclear. In this study, by analyzing previously published sequencing data of 1082 MTB Beijing isolates, we determined the genetic changes that were commonly present in modern Beijing strains but absent in ancient Beijing strains. These changes include 44 single-nucleotide polymorphisms (SNPs) and two short genomic deletions. Through bioinformatics analysis, we demonstrated that these genetic changes had high probability of functional effects. For example, 4 genes were frameshifted due to premature stop mutation or genomic deletions, 19 nonsynonymous SNPs located in conservative codons, and there is a significant enrichment in regulatory network for all nonsynonymous mutations. Besides, three SNPs located in promoter regions were verified to alter downstream gene expressions. Our study precisely defined the genetic features of modern Beijing strains and provided interesting clues for future researches to elucidate the mechanisms that underlie this sublineage’s successful expansion. These findings from the analysis of the modern Beijing sublineage could provide us a model to understand the dynamics of pathogenicity of MTB.
INTRODUCTION
Mycobacterium tuberculosis (MTB) isolates are currently classified into seven major lineages (lineage 1–7), and among them, lineage 2 is one of the most successful lineages with increasing prevalence in global population.Citation1 Lineage 2, also known as Beijing family, was first found to be dominant in South and East Asia.Citation2,Citation3 In the past two decades, Beijing strains caused a great concern because of the frequent associations with higher mutation rate, hypervirulence, immune evasion, treatment failures and drug resistance.Citation4 However, these associations or characteristics varied among different studies and were inconclusive, suggesting that genetic heterogeneity might exist within this family.Citation4,Citation5,Citation6,Citation7
Initially, Beijing strains were found highly homogenous based on genotyping data.Citation8 Study from Dick van Soolingen et al. suggested that they were recently selected by Bacille Calmette-Guerin (BCG) vaccination,Citation8 but recent studies found that the diversity within Beijing strains is higher than previously estimated and the expansion of Beijing strains is prior to the BCG vaccination.Citation1,Citation9,Citation10 Based on the existence or absence of IS6110 insertion(s) in the noise transfer function (NTF) region, Beijing family trains were further divided into modern (typical) and ancient (atypical) Beijing sublineages.Citation11 Of them, modern Beijing sublineage is the most prevalent Beijing sublineage in worldwide regions except in Japan and Korea.Citation2,Citation3,Citation5,Citation11 However, even in Japan, a rapid emergence and increasing prevalence of modern Beijing sublineage strains were reported.Citation12 The increasing prevalence of modern Beijing strains suggests that this sublineage might exhibit selective advantage over ancient Beijing sublineage. This selective advantage could be evaluated through various virulence-associated characters. For example, modern Beijing strains induced a lower level of proinflammatory cytokines (IL-1β, IFN-γ, IL-22) compared with ancient Beijing strains in peripheral blood mononuclear cells.Citation13 In both mice pulmonary infection and macrophage infection models, modern Beijing strains were more likely to exhibit highly virulent phenotypes than ancient Beijing strains.Citation14
Although modern Beijing strains have been a research hotspot in recent years, the genetic basis that contributes to their global prevalence is still unclear. As MTB lack horizontal gene transfer and recombination, the genomic evolution of MTB was characterized by stepwise accumulation of mutations.Citation15,Citation16 Identification of modern Beijing-specific genetic changes may provide important clues for understanding their phenotypic advantages. Modern Beijing strains are known to display missense alterations in three putative mutation repair genes (Rv3908, mutT2 and ogt) and these mutations were supposed to confer advantages of the rapid adaption to new environment as they might increase mutation rate.Citation6,Citation9 However, it is still not clear whether they play a role in the adaptation of modern Beijing strains as the influence of these mutations have not been validated yet.Citation4,Citation17 Thus, a comprehensive characterization of genetic features of modern Beijing strains is still needed. Previously, Anita C. Schurch et al have defined a minimal set of polymorphisms (51 single-nucleotide polymorphisms (SNPs)) for modern Beijing strains through typing 150 MTB strains with a subset of the SNPs they identified, and concluded that mutations in the regulatory network underlay its recent clonal expansion.Citation18 More recently, Matthias Merker et al. examined the characteristics of modern Beijing strains as 81 SNPs based on whole-genome sequencing of 110 globally collected MTB Beijing isolates.Citation19 The inconsistency of the two studies indicated that the variation was due to different sample sets they studied. Thus, investigating more representative isolates would lead to more accurate definition of modern Beijing genetic features. In recent years, a large number of whole-genome sequencing data of MTB Beijing strains has been published,Citation1,Citation10,Citation19,Citation20,Citation21 which provided the possibility to further illustrate evolutionary route of modern Beijing sublineage. In this study, taking advantage of large number of online available sequencing data of MTB Beijing strains, we defined the genetic features of modern Beijing sublineage more precisely and further demonstrated that these changes would probably cause functional effects.
MATERIALS AND METHODS
Genome sequencing data and SNPs/INDELs calling
Whole-genome sequencing data of MTB isolates were obtained from National Center for Biotechnology Information (NCBI) and European Nucleotide Archive (ENA. We used both SNP G-A in codon 176 of Rv2952 and SNP C→G in codon 20 of Rv2450c to identify Beijing strains as previously described.Citation5 Totally, whole-genome sequencing data of 1082 Beijing strains were included in our study and listed in Supplementary Table S1. The strains were from 15 countries with the major contribution from China and Russia (Supplementary Table S1). There were 193 strains from China, where Beijing strains originated and the highest genetic diversity was observed. Thus, we argue that the data set included in our study in geographically and genetically representative. The FASTQ format raw sequencing dat were processed by Scythe (https://github.com/ucdavis-bioinformatics/scythe) and Sickle (https://github.com/najoshi/sickle) for trimming adapter and low-quality bases, respectively. We discarded low-quality bases with Phred quality scores lower than 20. After mapping to the reference genome MTB H37Rv (GenBank accession NC_000962.3) with Burrows–Wheeler Aligner,Citation22 SNPs were called against the reference with a minimum depth of 20 folds coverage using SAMtools.Citation23 For insertions and deletions (INDELs) calling, we used VelvetCitation24 for de novo assembly and used BLATCitation25 to align assembled contigs with the reference genome MTB H37Rv. The INDELs between each sequenced isolate and the reference genome were then identified from the alignment file. Mobile genetic elements, repetitive sequences, PE/PPE and PE-PGRS gene families that might cause incorrect read alignment were excluded in this study.
Identify modern Beijing genetic features
A maximum likelihood (ML) tree was constructed in MEGA5Citation26 using the default parameters with the union of 55630 SNPs identified in the sequenced data and the robustness of the ML tree was validated by a 1000-time bootstrap test. Another four published Beijing strains genome data (CCDC5079, accession: NC_017523; CCDC5180, accession: NC_017522.1; X122, accession: CM001044; HN878, accession: CM001043; MTB 210, accession: ADAB00000000) were also included for a comprehensive view of Beijing family phylogenetic structure. The phylogenetic tree was visualized with Fig Tree (http://tree.bio.ed.ac.uk/software/figtree/). We applied two approaches to identify modern Beijing-specific mutations. First, we identified modern Beijing strains based on mutT2 G58R, ogt codon 12 (GGG-GGA) mutations and IS6110 insertion(s) in NTF region. Then we wrote a Perl script to identify the SNPs and INDELs that were uniformly presented in all modern Beijing strains but absent in closest ancient Beijing strains. Second, we reconstructed the ML phylogeny of Beijing strains based on the SNPs called from all strains in this study and further used MEGA5 to reconstruct the most recent common ancestor (MRCA) sequence of all modern Beijing strains (MRCA-modern) and also the MRCA sequence of the closest ancient Beijing node (MRCA-ancient. Through blast the two MRCA sequences, we identified modern Beijing-specific SNPs as the SNPs presented in MRCA-modern sequence against MRCA-ancient sequence.
Bioinformatics analysis
We downloaded gene annotation file from TBDB (http://www.tbdb.org/) and identified synonymous and nonsynonymous mutations based on the reference genome H37Rv. We download protein sequence from Uniprot and NCBI by choosing ‘Mycobacteria’ branch and perform PSI-BLASTCitation27 for each TB protein-coding gene. Then, we could get conservation score for each position of each TB gene, with score ranges from 0 to 4.36 with higher score indicating higher conservation. If the score is higher than 2.5, the corresponding site can be defined as a conservative site. For all nonsynonymous SNPs, we could detect whether it is located at a conservative position. We used SMART (http://smart.embl-heidelberg.de/) to identify protein domains and analyze the domain architectures. We performed functional enrichment analysis for the genes influenced by the modern Beijing nonsynonymous SNPs using annotation system from Tuberculist,Citation28 GO,Citation29 KEGGCitation30 and COG.Citation31 For each annotated gene set, we performed Fisher’s exact test to find out ones that were significantly over-represented in the genes with modern Beijing SNPs. The ChIP-Seq data sets were downloaded from TBDB. We used MEMECitation32 to predict PWM for each transcription factor (TF) and scan the whole genome to get potential TF binding site (TFBS). Then, we scanned the input SNP to find whether it located at TFBS and changed the binding potential of the corresponding TF. All SNPs were mapped to the TFBS at each position, and the TF binding potential score was calculated for the original TFBS and altered TFBS.
Luciferase report system assays
For the seven genes that were predicted to be affected by the mutations in TFBS, about 500 bp upstream sequences from start codon of these genes were amplified by PCR from both ancient Beijing (wild-type) and modern Beijing (mutant-type) strains, and then cloned into the pSMT3L-EGFP vector. A total of 14 plasmids were obtained and transferred into Mycobacterium smegmatis through electroporation. The M. smegmatis strains carrying these plasmids were cultured in 7H9 medium. Cultures (10 mL) of M. smegmatis were grown to OD600 = 0.6∼0.8 and then standardized to OD600 = 0.1. Luciferase activity was determined immediately after the collection of the cultures through the GloMax® 20/20 Luminometer (Promega). Three biological repeats were performed.
Ethic statement
All the whole-genome sequencing data analyzed were downloaded from online available resource (NCBI and ENA) and we did not generate new sequencing data in this study. Thus, there is no ethic/consent statement to make.
RESULTS
Genetic features of modern Beijing sublineage
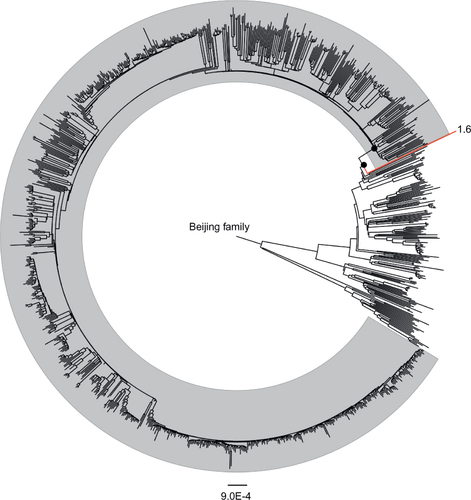
Whole-genome sequencing data of 1082 MTB Beijing isolates representing global diversity were downloaded from NCBI and ENA. Through mapping the sequencing reads to reference genome H37Rv, we called SNPs of each isolate against the H37Rv reference genome sequence. Among the 1082 Beijing isolates, 900 were determined to belong to modern Beijing sublineage by matching all three genetic markers (mutT2 G58R, ogt codon 12 GGG-GGA and IS6110 insertion in NTF region), and 181 were determined as ancient Beijing isolates in the absence of these genetic markers. One isolate named ‘1-6’ (sampled from Shanghai, China according to the original study) showed ambiguous characteristics according to the definitions. This isolate, although carrying mutT2 G58R mutation, was detected as wild type in ogt codon 12 and showed intact NTF region without IS6110 insertion. We reconstructed a maximum-likelihood phylogeny of the 1082 MTB Beijing isolates (). We applied two approaches to define the genetic features of modern Beijing sublineage. First, we identified the SNPs and insertions/deletions that were uniformly presented in modern Beijing strains but absent in the closest ancient Beijing branches. Second, we identified modern Beijing-specific SNPs through reconstructing the MRCA sequence of modern Beijing sublineage and compared it with the MRCA sequence of the closest ancient Beijing node (). Totally, 44 SNPs and 2 short genomic deletions were identified as modern Beijing-specific genetic features. Compared with the results from Matthias Merker et al.,Citation19 we excluded 43 SNPs that were not specific to modern Beijing strains but newly identified six SNPs and one genomic deletion. Comparing our results with the previous study by Schurch AC et al.,Citation18 we narrowed down the minimum set by excluding seven mutations and newly identified two short genomic deletions. The reason we narrowed down the minimum SNP set is that we included more ancient Beijing strains that were genetically closer to modern Beijing strains. The major contribution to the reduction of minimum SNP set was from the strain ‘1-6.’ This isolate showed closest distance to modern Beijing sublineage but was excluded from the expanded branch of modern Beijing sublineage (). The isolate ‘1-6’ harbored mutT2 G58R mutation but not ogt codon 12 mutation or IS6110 insertion in NTF region. Therefore, the mutations present in strain ‘1-6’ should not be modern Beijing-specific mutations and mutT2 G58R mutation was actually not strictly restricted to modern Beijing sublineage.
Figure 1 Maximum-likelihood phylogeny of 1082 MTB Beijing isolates. Strains shown in gray shadow represent modern Beijing sublineage while others belong to ancient Beijing sublineage. The two dark circles represent the MRCA sequences of modern Beijing strains and the closest ancient branch, respectively. Ancient Beijing strain 1-6 that harbored mutT2 G58R mutation but not IS6110 insertion in NTF region is marked in the phylogeny.
The 44 modern Beijing SNPs consist of 27 nonsynonymous (including one premature stop codon mutation), 13 synonymous and four intergenic mutations (Supplementary Table S2). The premature stop codon mutation in Rv2180c caused the coding protein truncated from 295 to 249 amino acids (). For the two short deletions, one is a single-nucleotide deletion in the overlap region of genes Rv2147c and Rv2148c that caused frameshift mutations for both genes. Rv2147c was shortened from 241 to 218 amino acids and Rv2148c had a frame shift alteration of the 5 tail amino acids (). The other was a 19-nucleotide deletion in gene Rv1730c, leading to the premature termination of Rv1730c with the coding protein length shortened from 517 to 418 amino acids, which likely results in inactivation of Rv1730c (). Among the four genes above, Rv1730c and Rv2147c were essential genes.Citation33 Rv1730c possibly codes a penicillin-binding protein, was involved in cell wall biosynthesis and may also act as a sensor of external penicillin.Citation33 Rv2147c was a core mycobacterial gene that is specific for mycobacteria and intracellular mycobacterial pathogens.Citation34 It is noteworthy that all the four genes mentioned above were located in cell wall or cell membrane. As for bacteria pathogens, the proteins in cell envelope are crucial to maintain the stability and integrity of cell and play an important role in virulence, host cell interaction and immune responses. Thus, these changes might have functional impacts on the cell envelope structure or host immunological recognition.
Table 1 Genes with significant alterations in modern Beijing strains
Potential functional changes revealed by bioinformatics analysis
Of the 27 genes carrying nonsynonymous mutations, we further performed conservative analysis and gene function enrichment analysis to predict their functional effects. Totally, 10 genes with nonsynonymous mutations encoded proteins located in cell wall or outer membrane (Supplementary Table S2), and these mutated genes were prone to frequent contact with host immune system. Conservative analysis showed that 18 of the 27 nonsynonymous SNPs were in the comparative conservative codons (Supplementary Table S3), suggesting that they might lead to functional changes of the related proteins. Among the genes with mutations in conservative codons, nine encoded enzymes and involved in metabolism or signal transduction (Supplementary Table S3). Gene function enrichment analysis revealed that the genes with nonsynonymous mutations were mainly enriched in regulatory protein network (, P = 0.029), which was also revealed by Schurch AC et al. earlier.Citation18 In modern Beijing strains, PknA has two adjacent amino changes (Q369R and Q370P) in the low-complexity region of outer membrane domain and both changes altered the property of the amino acids. Among these affected regulatory proteins, PknA is widely used to transduce extracellular signals into appropriate intracellular responses and involved in numerous cellular processes, and the alteration of sensor domain might lead to a difference in response to external stimulus.Citation35,Citation36 Rv0890c and Rv2488c are both LuxR family regulators involved in MTB dormancy.Citation37,Citation38 Previous study showed that the loss of a LuxR family regulator Rv0386 (an adenylate cyclase) decreased immunopathology in animal tissues and bacterial survival.Citation39 Coding changes in transcriptional regulators are considered to be rare because their multifunctional roles and a small change would produce widespread detrimental effects, and mutations in regulatory networks were also thought to involve in bacteria’s adaptation to host.Citation40 Thus, the significant enrichment of regulatory network proteins might be a reflection of modern Beijing sublineage’s adaptive microevolution.
Table 2 Modern Beijing-specific mutations that are enriched in the gene category of regulatory network and other mutations with potential functional effects
Modern Beijing SNPs altered downstream genes expression
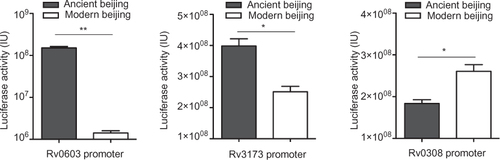
Among the 44 modern Beijing-specific SNPs, five were predicted to locate in the TFBSs and might alter the expression of downstream genes (Supplementary Table S4). We further experimentally investigated whether these SNPs would affect downstream gene expression through luciferase report system in M. smegmatis. Three SNPs were verified to have impacts on the expression of downstream genes (). Among them, mutation 16 A-C in the promoter of Rv0603 caused ∼100-fold downregulation. Rv0603 possibly codes an exported protein but its function was unknown.Citation41,Citation42 Thus, it is hard to infer the biological changes due to low expression of Rv0603. Mutation in predicted promoter of Rv3173c led to 1.4-fold decreased regulation. However, this small change could amplify into great influence as Rv3173c codes a TetR/AcrR family transcriptional regulator that regulates a large number of genes.Citation43 Rv0308 probably codes a conserved integral membrane protein and mutation in the predicted promoter of Rv0308 led to ∼1.6-fold increase of expression. The other two mutations did not have significant impact on the downstream genes’ expression. These results confirmed the functional effects of modern Beijing SNPs, but due to the poor knowledge of these genes’ functions, we could not make an inference for the biological effects of these alterations. Thus, it will be interesting to verify whether these alterations would contribute to some phenotype changes. The other three SNPs that did not change the downstream genes’ expression are shown in Supplementary Figure S1.
Figure 2 Modern Beijing-specific SNPs alter the expression level of downstream genes. The promoters with modern Beijing-specific mutations have been tested for transcriptional activity against promoters without respective mutations through luciferase report system in M. smegmatis. Data presented are mean values from three independent experiments. Error bars, 95% confidence intervals.
DISCUSSION
A recent study based on phylogeographic and coalescent analyses of large number of Beijing strains indicated that Beijing family emerged around 30 000 years ago in southern East Asia, which was accompanied with the early colonization by modern humans in this area.Citation10 After that, Beijing strains branched into several major sublineages during the coevolution with human beings in Asia. Among them, modern Beijing sublineage was the latest branch but experienced the most significant expansion during the neolithic demographic transition (NDT, 6000–7000 years ago), suggesting that this sublineage had successfully adapted to the environment change of increasing human population densities during NDT.Citation10 Thus, identification of genetic difference between modern Beijing and other Beijing sublineages would help us understand the pathogenicity evolution of MTB. In this study, through analyzing large number of published whole-genome sequencing data of MTB, we reconstructed the most representative phylogeny of Beijing strains and characterized the genetic features that were restricted to modern Beijing sublineage. We argue that the narrowing down of modern Beijing-specific SNPs in our study compared with previous studies is more precise because we included a lot more representative Beijing strains for analysis.
We performed several analysis or experiments to investigate the functional effects of modern Beijing genetic features. Through bioinformatics analysis, we found that three genes were truncated and one gene was frameshifted. Truncations of genes usually lead to loss of gene function, but due to the poor annotation of these genes, we could not speculate the biological influence directly. Yet, all the four genes were located in cell membrane or cell wall, genes of which category are frequently associated with maintaining cell envelop integrity or immunological recognition. Thus, revealing these genes’ function would help us understand the microevolution of MTB modern Beijing strains. Nonsynonymous mutations in conservative codons might lead to changes in the enzyme activity or structure of encoded proteins. Our conservative analysis of the 27 nonsynonymous mutations demonstrated that 18 of them were located in conservative sites, which further supported the functional effects of modern Beijing genetic features. Remodeling of global regulatory networks is a key route for bacterial pathogen adaptation to the fluctuations of host environment;Citation44 thus, the significant enrichment in regulatory protein category of modern Beijing-specific nonsynonymous mutations might be an indicator of pathogen microevolution.
Besides, several other mutations that could affect the virulence of modern Beijing strains are also listed. Modern Beijing strains have a nonsynonymous mutation at a conservative site in secA2. MTB has two SecA proteins, SecA1 and SecA2; SecA1 handles ‘housekeeping’ export while SecA2 exports a specific subset of virulence factors and is necessary to elude the oxidative attack of macrophages.Citation45,Citation46 Another conservative-site mutation was observed in sseA, which codes a putative thiosulfate sulfurtransferase, and a study showed that mutation in this gene could lead to enhanced growth in macrophages relative to in vitro growth.Citation47 More recently, de Keijzer J et al. further found that SseA was among the four proteins that were differentially regulated between ancient and modern Beijing sublineages.Citation48 Mutation in glcB was at a highly conservative site and this gene encodes a malate synthase. GlcB takes part in the glyoxylate shunt and has been implicated as a virulence factor, which was also important for MTB survival under adverse conditions, such as low oxygen, nonreplicative states and the intracellular environment.Citation49 Thus, it will be interesting to verify whether these mutations would cause some changes on protein functions.
However, our study still has several limitations. First, due to the lower prevalence of ancient Beijing strains, the number of isolates analyzed belonging to the modern sublineage is much higher than those from the ancient Beijing sublineage. Thus, if more ancient Beijing strains that showed even closer relationship to modern Beijing sublineage were sequenced, the minimum set of modern Beijing-specific genetic features might be further reduced. Second, the approach we used to validate the change of downstream genes’ expression was through luciferase report system in M. smegmatis. Although this approach was commonly accepted and used for this purpose, we failed to further confirm these findings in MTB isolates due to the requirement for biosafety level 3 lab to culture MTB and extract RNA.
In conclusion, we identified the genetic features that were restricted to modern Beijing sublineage and revealed that they had functional effects. Through characterizing the modern Beijing-specific genetic changes, our findings provided new clues to elucidate the successful expansion or hypervirulence of MTB modern Beijing strains. As Beijing family is one of the most successful lineages of global MTB strains, these investigations into modern Beijing sublineage could provide us a model to understand the dynamics of pathogenicity in MTB.
Tuberculosis: mapping the evolution of virulence
Genomic analysis reveals mutations that could contribute to enhanced virulence in a widely prevalent subtype of tuberculosis. The ‘Beijing family’ of Mycobacterium tuberculosis has existed for over 30 000 years, but there is also a more recently evolved ‘modern sublineage’ that spreads more aggressively in human populations. Researchers led by Qian Gao at Fudan University analyzed genomic data from over 1 000 M. tuberculosis isolates to identify dozens of sequence changes specific to this sublineage. Some affect production of proteins on the bacterial surface, which could alter interactions with host cells and the immune system. Others were linked to proteins that control regulatory networks with far-reaching effects, which could shape the extent to which M. tuberculosis can adapt to environmental changes. More thorough characterization of these genomic changes could offer valuable insights into this pathogen’s survival strategies.
Supplementary Table S1
Download MS Excel (48.2 KB)Supplementary Table S2
Download MS Excel (881.9 KB)Supplementary Table S3
Download MS Excel (879.4 KB)Supplementary Table S4
Download MS Excel (878.4 KB)Supplementary Figure S1
Download PDF (253.8 KB)This work was supported by the Natural Science Foundation of China (91231115 and 31301033) and China Postdoctoral Science Foundation (2012M52082). This work was also partially supported by the National Institutes of Health (USA) through the Fogarty International Center (D43TW007887).
Related Research Data
- ComasI,CoscollaM,LuoTet al.Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans.Nat Genet2013; 45:1176–1182.
- MokrousovI,LyHM,OttenTet al.Origin and primary dispersal of the Mycobacterium tuberculosis Beijing genotype: clues from human phylogeography.Genome Res2005; 15:1357–1364.
- BifaniPJ,MathemaB,KurepinaNE,KreiswirthBN.Global dissemination of the Mycobacterium tuberculosis W-Beijing family strains.Trends Microbiol2002; 10:45–52.
- ParwatiI,van CrevelR,van SoolingenD.Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains.Lancet Infect Dis2010; 10:103–11.
- YangC,LuoT,SunGet al.Mycobacterium tuberculosis Beijing strains favor transmission but not drug resistance in China.Clin Infect Dis2012; 55:1179–1187.
- LanNT,LienHT,Tung leB,BorgdorffMW,KremerK,van SoolingenD.Mycobacterium tuberculosis Beijing genotype and risk for treatment failure and relapse, Vietnam.Emerg Infect Dis2003; 9:1633–1635.
- WerngrenJ,HoffnerSE.Drug-susceptible Mycobacterium tuberculosis Beijing genotype does not develop mutation-conferred resistance to rifampin at an elevated rate.J Clin Microbiol2003; 41:1520–1524.
- van SoolingenD,QianL,de HaasPEet al.Predominance of a single genotype of Mycobacterium tuberculosis in countries of east Asia.J Clin Microbiol1995; 33:3234–3238.
- MestreO,LuoT,Dos VultosTet al.Phylogeny of Mycobacterium tuberculosis Beijing strains constructed from polymorphisms in genes involved in DNA replication, recombination and repair.PLoS One2011; 6:e16020.
- LuoT,ComasI,LuoDet al.Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese.Proc Natl Acad Sci USA2015; 112:8136–8141.
- MokrousovI,NarvskayaO,OttenTet al.Phylogenetic reconstruction within Mycobacterium tuberculosis Beijing genotype in northwestern Russia.Res Microbiol2002; 153:629–637.
- IwamotoT,FujiyamaR,Yoshida S WadaT,ShiraiC,KawakamiY.Population structure dynamics of Mycobacterium tuberculosis Beijing strains during past decades in Japan.J Clin Microbiol2009; 47:3340–3343.
- van LaarhovenA,MandemakersJJ,KleinnijenhuisJet al.Low induction of proinflammatory cytokines parallels evolutionary success of modern strains within the Mycobacterium tuberculosis Beijing genotype.Infect Immun2013; 81:3750–3756.
- RibeiroSC,GomesLL,AmaralEPet al.Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage.J Clin Microbiol2014; 52:2615–2624.
- DyeC.Doomsday postponed? Preventing and reversing epidemics of drug-resistant tuberculosis.Nat Rev Microbiol2009; 7:81–87.
- ZainuddinZF,DaleJW.Does Mycobacterium tuberculosis have plasmids?Tubercle1990; 71:43–49.
- MorelandNJ,CharlierC,DingleyAJ,BakerEN,LottJS.Making sense of a missense mutation: characterization of MutT2, a Nudix hydrolase from Mycobacterium tuberculosis, and the G58R mutant encoded in W-Beijing strains of M. tuberculosis.Biochemistry2009; 48:699–708.
- SchurchAC,KremerK,WarrenRMet al.Mutations in the regulatory network underlie the recent clonal expansion of a dominant subclone of the Mycobacterium tuberculosis Beijing genotype.Infect Genet Evol2011; 11:587–597.
- MerkerM,BlinC,MonaSet al.Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage.Nat Genet2015; 47:242–249.
- CasaliN,NikolayevskyyV,BalabanovaYet al.Evolution and transmission of drug-resistant tuberculosis in a Russian population.Nat Genet2014; 46:279–286.
- FarhatMR,ShapiroBJ,KieserKJet al.Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis.Nat Genet2013; 45:1183–1189.
- LiH,DurbinR.Fast and accurate short read alignment with Burrows-Wheeler transform.Bioinformatics2009; 25:1754–1760.
- LiH,HandsakerB,WysokerAet al.The Sequence Alignment/Map format and SAMtools.Bioinformatics2009; 25:2078–2079.
- ZerbinoDR,BirneyE.Velvet: algorithms for de novo short read assembly using de Bruijn graphs.Genome Res2008; 18:821–829.
- KentWJ.BLAT—the BLAST-like alignment tool.Genome Res2002; 12:656–664.
- TamuraK,PetersonD,PetersonN,StecherG,NeiM,KumarS.MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods.Mol Biol Evol2011; 28:2731–2739.
- AltschulSF,MaddenTL,SchafferAAet al.Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.Nucleic Acids Res1997; 25:3389–3402.
- LewJM,KapopoulouA,JonesLM,ColeST.TubercuList–10 years after.Tuberculosis (Edinb)2011; 91:1–7.
- AshburnerM,BallCA,BlakeJAet al.Gene ontology: tool for the unification of biology. The Gene Ontology Consortium.Nat Genet2000; 25:25–29.
- KanehisaM,GotoS.KEGG: kyoto encyclopedia of genes and genomes.Nucleic Acids Res2000; 28:27–30.
- TatusovRL,GalperinMY,NataleDA,KooninEV.The COG database: a tool for genome-scale analysis of protein functions and evolution.Nucleic Acids Res2000; 28:33–36.
- BaileyTL,WilliamsN,MislehC,LiWW.MEME: discovering and analyzing DNA and protein sequence motifs.Nucleic Acids Res2006; 34:W369–W373.
- SassettiCM,BoydDH,RubinEJ.Genes required for mycobacterial growth defined by high density mutagenesis.Mol Microbiol2003; 48:77–84.
- MarmiesseM,BrodinP,BuchrieserCet al.Macro-array and bioinformatic analyses reveal mycobacterial ‘core’ genes, variation in the ESAT-6 gene family and new phylogenetic markers for the Mycobacterium tuberculosis complex.Microbiology2004; 150:483–496.
- ThakurM,ChakrabortiPK.Ability of PknA, a mycobacterial eukaryotic-type serine/threonine kinase, to transphosphorylate MurD, a ligase involved in the process of peptidoglycan biosynthesis.Biochem J2008; 415:27–33.
- ThakurM,ChabaR,MondalAK,ChakrabortiPK.Interdomain interaction reconstitutes the functionality of PknA, a eukaryotic type Ser/Thr kinase from Mycobacterium tuberculosis.J Biol Chem2008; 283:8023–8033.
- ZengLR,XieJP.Molecular basis underlying LuxR family transcription factors and function diversity and implications for novel antibiotic drug targets.J Cell Biochem2011; 112:3079–3084.
- SantosCL,Correia-NevesM,Moradas-FerreiraP,MendesMV.A walk into the LuxR regulators of Actinobacteria: phylogenomic distribution and functional diversity.PLoS One2012; 7:e46758.
- AgarwalN,LamichhaneG,GuptaR,NolanS,BishaiWR.Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase.Nature2009; 460:98–102.
- DamkiaerS,YangL,MolinS,JelsbakL.Evolutionary remodeling of global regulatory networks during long-term bacterial adaptation to human hosts.Proc Natl Acad Sci USA2013; 110:7766–7771.
- BecqJ,GutierrezMC,Rosas-MagallanesVet al.Contribution of horizontally acquired genomic islands to the evolution of the tubercle bacilli.Mol Biol Evol2007; 24:1861–1871.
- MalenH,BervenFS,FladmarkKE,WikerHG.Comprehensive analysis of exported proteins from Mycobacterium tuberculosis H37Rv.Proteomics2007; 7:1702–1718.
- CamusJC,PryorMJ,MedigueC,ColeST.Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv.Microbiology2002; 148:2967–2973.
- DamkiaerS,YangL,MolinS,JelsbakL.Evolutionary remodeling of global regulatory networks during long-term bacterial adaptation to human hosts.Proc Natl Acad Sci USA2013; 110:7766–7771.
- FeltcherME,GibbonsHS,LigonLS,BraunsteinM.Protein export by the mycobacterial SecA2 system is determined by the preprotein mature domain.J Bacteriol2013; 195:672–681.
- HouJM,D'LimaNG,RigelNWet al.ATPase activity of Mycobacterium tuberculosis SecA1 and SecA2 proteins and its importance for SecA2 function in macrophages.J Bacteriol2008; 190:4880–4887.
- RengarajanJ,BloomBR,RubinEJ.Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages.Proc Natl Acad Sci USA2005; 102:8327–8332.
- de KeijzerJ,de HaasPE,de RuAH,van VeelenPA,van SoolingenD.Disclosure of selective advantages in the “modern” sublineage of the Mycobacterium tuberculosis Beijing genotype family by quantitative proteomics.Mol Cell Proteomics2014; 13:2632–2645.
- SmithCV,HuangCC,MiczakA,RussellDG,SacchettiniJC,Höner zu BentrupK.Biochemical and structural studies of malate synthase from Mycobacterium tuberculosis.J Biol Chem2003; 278:1735–1743.