
Abstract
Enterovirus 71 induces dsRNA/PKR-dependent cytoplasmic redistribution of GRP78/BiP to promote viral replication
GRP78/BiP is an endoplasmic reticulum (ER) chaperone protein with the important function of maintaining ER homeostasis, and the overexpression of GRP78/BiP alleviates ER stress. Our previous studies showed that infection with enterovirus 71 (EV71), a (+)RNA picornavirus, induced GRP78/BiP upregulation; however, ectopic GRP78/BiP overexpression in ER downregulates virus replication and viral particle formation. The fact that a virus infection increases GRP78/BiP expression, which is unfavorable for virus replication, is counterintuitive. In this study, we found that the GRP78/BiP protein level was elevated in the cytoplasm instead of in the ER in EV71-infected cells. Cells transfected with polyinosinic–polycytidylic acid, a synthetic analog of replicative double-stranded RNA (dsRNA), but not with viral proteins, also exhibited upregulation and elevation of GRP78/BiP in the cytosol. Our results further demonstrate that EV71 infections induce the dsRNA/protein kinase R-dependent cytosolic accumulation of GRP78/BiP. The overexpression of a GRP78/BiP mutant lacking a KDEL retention signal failed to inhibit both dithiothreitol-induced eIF2α phosphorylation and viral replication in the context of viral protein synthesis and viral titers. These data revealed that EV71 infection might cause upregulation and aberrant redistribution of GRP78/BiP to the cytosol, thereby facilitating virus replication.
Emerging Microbes and Infections (2016) 5, e23; doi:10.1038/emi.2016.20; published online 23 March 2016
INTRODUCTION
Enterovirus 71 (EV71) belongs to the enterovirus genus of Picornaviridae, which infect mostly infants or children younger than five years and may cause hand, foot and mouth disease.Citation1 Neurological complications, including encephalitis and aseptic meningitis, are frequently manifested upon severe infection.Citation2 EV71 particles lack an envelope and consist of an icosahedral capsid and a 7.4-kb single positive-stranded RNA. The viral RNA encodes a polyprotein that is subsequently cleaved by viral proteases (2Apro, 3Cpro and 3CDpro) to produce individual viral proteins: structural proteins (VP4, VP2, VP3 and VP1) constitute the capsid shell, whereas nonstructural proteins (2A, 2B, 2C, 2BC, 3A, 3B, 3AB, 3C, 3D and 3CD) are associated with viral replication. In addition, a double-stranded RNA (dsRNA) intermediate produced during virus replication has been shown to trigger antiviral responses or alter cellular processes.Citation3 dsRNA-dependent protein kinase R (PKR) is a dsRNA-binding protein that consists of an N-terminal dsRNA-binding domain and a C-terminal kinase domain.Citation4 After activation by dsRNA, PKR can phosphorylate eIF2α, which results in the global arrest of protein synthesis. However, some genes that are involved in the integrated stress response (ISR), such as ATF4 and CHOP, are selectively activated under eIF2α phosphorylation to relieve stress or regulate cell death.Citation5
The molecular chaperone 78-kDa glucose-regulated protein (GRP78), also referred to as binding immunoglobulin protein (BiP) or heat-shock 70-kDa protein A5 (HSPA5), is expressed primarily in the endoplasmic reticulum (ER).Citation6 GRP78/BiP contains an N-terminal ATPase domain, a C-terminal polypeptide-binding domain, and a highly helical C-terminal tail with a KDEL ER retention signal.Citation7, Citation8, Citation9, Citation10 The function of GRP78/BiP has previously been shown differ by subcellular localization.Citation11 For example, the GRP78/BiP expressed on the cell surface is reported to serve as a receptor for cancer cells to modulate cellular signaling pathways, such as mitogen-activated protein kinase (p38 MAPK), extracellular-signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinases (JNKs), and phosphoinositide 3-kinase (PI3K).Citation12, Citation13 In addition, a cytosolic GRP78/BiP isoform (GRP78va) may be critical for cell viability.Citation14 These findings unveiled a potent antiapoptotic role for GRP78/BiP; therefore, it might be an ideal target for cancer therapy.
Dengue virus infection induces an increase in BiP78/BiP on the cell surface, where it acts as a receptor for virus binding and entry.Citation15 In addition, the interaction between hepatitis B virus (HBV) precore protein and GRP78/BiP causes the redistribution of GRP78/BiP from the ER to the cytoplasm, which may account for persistent HBV infection.Citation16 GRP78/BiP is a well-known monitor of ER stress and has fundamental roles in the unfolded-protein response (UPR).Citation17 Under unstressed conditions, BiP/GRP78 associates with ER stress sensor proteins, for example, inositol-requiring protein 1 (IRE1), protein kinase RNA-activated (PKR)-like ER kinase (PERK) and activating transcription factor 6 (ATF6), to inactivate the sensor proteins. Under stressed conditions, the accumulation of unfolded and misfolded proteins recruits GRP78/BiP, which dissociates sensor proteins from GRP78/BiP and activates the downstream signal transduction cascade to rescue ER homeostasis. An increase in GRP78/BiP in the ER compartment has often been used as evidence of UPR induction. Many viruses manipulate ER stress or UPR to benefit their own replication,Citation18 including Japanese encephalitis virus, hepatitis C virus and dengue viruses.Citation19, Citation20, Citation21, Citation22 Our previous findings demonstrated that EV71 activated and modulated the UPR for viral replication.Citation23 The exogenous expression of GRP78/BiP attenuated EV71-induced ER stress and reduced viral RNA expression and infectious particle formation. Although these results have highlighted that ER stress or the modulated UPR promotes virus replication, the mechanism by which virus subverts GRP78/BiP, whose ectopic overexpression is disadvantageous to the virus, has not yet been identified. Therefore, we hypothesized that GRP78/BiP might lose its function of mitigating ER stress upon EV71 infection and explored the modulation of GRP78/BiP in virus-infected cells.
MATERIALS AND METHODS
Cell culture and infection
Human rhabdomyosarcoma (RD) and human embryonic kidney (HEK)293T cells were obtained from American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) (JRH Biosciences, Lenexa, KS, USA) at 37 °C in a 5% CO2-humidified environment. The EV71 (TW/2231/98, genotype C) infectious clone was generously provided by Dr Mei-Shang Ho of Academia Sinica, Taiwan. The virus was generated from the infectious clone and propagated in the RD cells, and the virus titer was determined by a plaque assay with RD cells.Citation24 UV-inactivated EV71 (UV-EV71) was prepared as described previously.Citation25 Viral infection was carried out by preadsorbing EV71 at a multiplicity of infection (MOI) of 10 for 1 h (–1 to 0 h post infection (p.i.)) at 37 °C, and the unbound virus was aspirated and washed with phosphate-buffered saline (PBS). The cells were then cultured in DMEM containing 2% FBS and harvested at the indicated times p.i.
Reagents and antibodies
Dithiothreitol (DTT), p38 MAPK inhibitor SB203580, Q-VD-OPh, anti-Flag antibody, dimethyl sulfoxide (DMSO) and polyinosinic–polycytidylic acid (poly(I:C)) were purchased from Sigma-Aldrich (St Louis, MO, USA). The JNK inhibitor SP600125 was purchased from BioVision (Milpitas, CA, USA). Staurosporine was purchased from Cell Signaling Technology (Danvers, MA, USA). Mouse anti-PKR antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-p-PKR, anti-KDELR, anti-Bax and anti-Calnexin antibodies were purchased from GeneTex (Irvine, CA, USA). Mouse anti-GRP78 and anti-GAPDH antibodies were obtained from BD Transduction (San Diego, CA, USA) and Abnova (Taipei, Taiwan), respectively. Rabbit polyclonal antibodies against the EV71 3A and 2C proteins were prepared as described previously.Citation26 The monoclonal antibody against EV71 3D was kindly provided by Dr Shin-Ru Shih of Chang Gung University, Taoyuan.Citation27 The anti-mouse IgG (H+L) and anti-rabbit IgG (H+L) antibodies were purchased from Millipore (Billerica, MA, USA). Mouse anti-HA antibody was purchased from Roche (Basel, Switzerland). The Plasma Membrane Protein Extraction Kit (ab65400) was purchased from Abcam (Cambridge, UK).
Subcellular fractionation
Cells were washed with PBS and resuspended in Bud Buffer (38 mM potassium aspartate, 38 mM potassium gluconate, 20 mM MOPS, 5 mM reduced glutathione, 5 mM sodium carbonate, 2.5 mM magnesium sulfate, pH 7.2) containing protease inhibitors (Roche). All chemicals were purchased from Sigma-Aldrich unless otherwise stated. The samples were homogenized with a cell cracker (HGN Laboratory Equipment, Heidelberg, Germany), and the resulting homogenates were centrifuged at 1000g for 10 min at 4 °C. The postnuclear supernatant (PNS) was loaded into the top of a step OptiPrep (Sigma-Aldrich) gradient (10, 20, 30 and 40% discontinuously), followed by centrifugation at 50000g for 18 h at 4 °C in a SW 55 Ti rotor (Beckman, Columbia, MD, USA). Fractions (0.3 mL each) were collected from the top of the gradient, and 40 μL of each fraction was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotting. For cytosolic/microsomal fractionation, the PNSs were centrifuged at 7000g for 10 min at 4 °C. The mitochondrial-rich pellet was discarded, and 300 μg of supernatant was spun at 65 000g for 45 min at 4 °C in a Beckman TLA 100.2 rotor to obtain cytosolic supernatant and a microsome-rich pellet. The microsome-rich pellet was dissolved in Bud Buffer.
Secretion assay
The cells were cultured in DMEM without FBS in a 10-cm culture dish and infected with EV71/2231 for 1 h. Next, they were washed with PBS to remove unbound virus and maintained in DMEM supplemented with 2% FBS for 2 h. The cells were then washed again with PBS and cultured in DMEM only for 6 h. After incubation, the cell culture medium and attached cells were processed separately as described below. The cell culture medium was carefully transferred to 10-kDa Amicon centrifugation filter units (Millipore) and centrifuged at 8000g for 45 min at 4 °C. The retentate was resuspended in lysis buffer (1% Triton X-100, 50 mM sodium chloride, 1 mM EDTA, 1 mM EGTA, 20 mM sodium fluoride, 20 mM sodium pyrophosphate, 1 mM sodium orthovanadate in 20 mM Tris-HCl, pH 7.4) containing a protease inhibitor cocktail (Roche) and then collected as the supernatant fraction. The attached cells were washed with PBS, resuspended in lysis buffer, and centrifuged at 17 900g for 10 min at 4 °C. The supernatant was collected as the whole-cell lysate.
Plasmid construction, siRNA and transfection
Full-length GRP78/BiP containing silent mutations in the siRNA target regions was cloned by polymerase chain reaction (PCR) using pcDNA3.1-GRP78FL-myc-His B+ as a templateCitation23 and the following primers: 5′-AAG CTG GCT AGC ATG GAG GAG GAG GAC AAG AAG GAG-3′, 5′-TCT TCG AGT GAC AGC AGA TGA CAA GGG TAC AG-3′, 5′-CTG TAC CCT TGT CAT CTG CTG TCA CTC GAA G-3′ and 5′-GAC GGA TAT CAG CAA CTC ATC TTT TTC TGC TGT ATC CTC-3′. GRP78_ΔKDEL was amplified using a different reverse primer 5′-GAC GGA TAT CAG TTC TGC TGT ATC CTC TTC ACC AGT TGG-3′. Both cDNAs were inserted into AS3W-puro-cFlag with NheI and EcoRV restriction enzymes to generate AS3W-GRP78_WT-cFlag and AS3W-GRP78_ΔKDEL-cFlag constructs. Full-length EV71 2B, 2C, 2BC, 3A, 3AB, 3D and 3CD were amplified from the infectious cDNA clone (TW/2231/98) by PCR and cloned into the NheI and EcoRI sites of AS3W-puro-cFlag. The primers for 2B were 5′-AAG CTG GCT AGC ATG GGC GTG TCT GAT TAC ATT AAA G-3′ and 5′-ATC GAT GAA TTC GTC TGC TTC TGA GCC ATC G-3′. The primers for 2C were 5′-AAG CTG GCT AGC ATG AGT GCC TCT TGG TTA AAG-3′ and 5′-CGA CTG AAT TCG TTT GGA AAA GAG CTT CAA TG-3′. The primers for 2BC were 5′-AAG CTG GCT AGC ATG GGC GTG TCT GAT TAC ATT AAA G-3′ and 5′-CGA CTG AAT TCG TTT GGA AAA GAG CTT CAA TG-3′. The primers for 3A were 5′-GCA TGG CTA GCA TGG GAC CCC CTA AAT TTA G-3′ and 5′-ATC GAT GAA TTC GTT TGA AAA CCG GCG AAC AAC-3′. The primers for 3AB were 5′-GCA TGG CTA GCA TGG GAC CCC CTA AAT TTA G-3′ and 5′-ATC GAT GAA TTC GTC TGC ACA GTT GCC GTG C-3′. The primers for 3D were 5′-AAG CTG GCT AGC ATG GGT GAG ATC CAA TGG ATG-3′ and 5′-CGA CTG AAT TCG TAA ACA ATT CGA GCC AAT TTC-3′. The primers for 3CD were 5′-GAT TAG CTA GCA TGG GGC CGA GCT TGG ACTT C-3′ and 5′-CGA CTG AAT TCG TAA ACA ATT CGA GCC AAT TTC-3′. Plasmids expressing 2Apro and 3Cpro were cloned into pcDNA3.1-mycHis(−).Citation28 Scrambled siRNA (medium GC of Stealth negative control duplex), siRNA against EIF2AK2 (PKR): 5′-UUU ACU UCA CGC UCC GCC UUC UCG U-3′ and siRNA against GRP78/BiP: 5′-UAC CCU UGU CUU CAG CUG UCA CUC G-3′ were purchased from Invitrogen (Carlsbad, CA, USA). The siRNA and constructs were transfected into designated cells using Lipofectamine 2000 (Invitrogen) as described by the manufacturer. The pKH3-KDELR plasmid expressing human KDEL receptor was kindly shared by Dr Guanghui Wang.Citation29
RNA extraction and quantitative PCR analysis
Total RNA was extracted from cells using TRIzol (Invitrogen). After DNase I treatment, 1 μg of total RNA was reverse-transcribed using the M-MLV reverse transcriptase system (Invitrogen). Quantitative PCR was performed using the StepOnePlus Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) with the following specific primer pairs: BiP forward primer 5′-CTC AAC ATG GAT CTG TTC CG-3′ and reverse primer 5′-CCA GTT GCT GAA TCT TTG GA-3′; GAPDH forward primer 5′-TGC ACC ACC AAC TGC TTA GC-3′ and reverse primer 5′-GGC ATG GAC TGT GGT CAT GAG-3′. The target genes were then amplified under the following conditions: 50 °C for 2 min, 95 °C for 10 min, 50 cycles at 95 °C for 15 s and 60 °C for 1 min. To quantify the changes in target gene expression, the comparative Ct (ΔΔCt) method was used to calculate relative fold changes normalized to the GAPDH control. Data are expressed as the mean±s.d. and were analyzed with two-tailed Student’s t-tests. P<0.05 was considered significant.
SDS–PAGE and western blotting assay
The protein concentration was measured with a protein assay kit (Bio-Rad, Richmond, CA, USA). Equal amounts of cellular protein were analyzed by SDS–PAGE and transferred to polyvinylidene fluoride membranes. The membranes were immunoblotted with specific primary antibodies followed by horseradish peroxidase (HRP)-conjugated secondary antibodies and developed with Immobilon Western HRP Chemiluminescence Substrate (Millipore).
GRP78/BiP rescue experiments
RD cells were transfected with either scrambled or GRP78 siRNA for three days. Then, AS3W-puro-cFlag (empty vector), AS3W-GRP78_WT-cFlag or AS3W-GRP78_ΔKDEL-cFlag construct was transfected into cells for another 24 h. After infecting with EV71 at an MOI of 10 for 6 h, the cell lysates were prepared for western blotting. The viral progeny inside the cells and in the culture medium were pooled for a plaque assay.Citation24 Data are expressed as the mean±s.d. and were analyzed with two-tailed Student’s t-tests. *** indicates P<0.001.
RESULTS
EV71 infection induces redistribution of GRP78/BiP
We previously showed that EV71 infection induced and modified the UPR.Citation23 The ER stress marker GRP78/BiP was upregulated at the protein level in response to viral infection. Because GRP78/BiP can target different subcellular compartments in response to stimuli,Citation11 we investigated the cellular localization of GRP78/BiP following EV71 infection by fractionating PNS obtained from mock-infected, EV71-infected or DTT (ER stress inducer)-treated RD cells on a step OptiPrep gradient (). Two main pools of GRP78/BiP were detected in the mock control gradient: one coincided with calnexin (fractions 6–9, designated as heavier fractions), an ER transmembrane protein and the other was present in the lighter fractions (fractions 2–4). We found that in DTT-treated cells, GRP78/BiP mostly cofractionated with calnexin in the heavier fractions (fractions 5–8). However, upon virus infection, the majority of GRP78/BiP was found in the lighter fractions (fractions 2–4), whereas viral nonstructural proteins 2C and 3A strongly cosedimented with calnexin (fractions 6–8). We determined the differential subcellular GRP78/BiP distribution in EV71-infected and DTT-treated RD cells (). We further confirmed the results with a different subcellular fractionation approach in which cytosol- and microsome-rich fractions were collected by differential centrifugation and analyzed by western blotting. Antibodies against calnexin and GAPDH were used to confirm the fractionation process. As expected, calnexin was expressed in the microsomal fraction, whereas GAPDH was exclusively found in the cytosol ). The total protein expression of GRP78/BiP was upregulated in EV71-infected RD cells (lane 1 versus 2, ), but its levels were elevated in the cytosol (lane 3 versus 4) and almost unchanged in the microsomal fractions (lanes 5 and 6, ). Conversely, GRP78/BiP was upregulated by DTT, and more GRP78/BiP expression was observed in the microsome-rich fraction (lanes 11 and 12, ). The same phenomenon was observed in HEK293T cells (), implying that the aberrant redistribution of GRP78/BiP to the cytosol was independent of cell type. Because a fraction of the GRP78/BiP cellular pool can be secreted from cells upon viral infectionCitation30 or anchored to the cell surface in some cancer cells,Citation11 we next examined the ability of EV71 infection to result in the secretion of GRP78/BiP or cell surface localization in addition to its effect on cytoplasmic redistribution. To detect cell surface GRP78/BiP, plasma membrane proteins from RD cells mock-infected or infected with EV71 (MOI of 10) were collected and extracted 6 h p.i. The extracts were resolved using SDS–PAGE and then analyzed with a western blot. As shown in , virus infection did not alter the cell surface expression level of GRP78/BiP compared with the mock-infected control (lanes 3 and 4, ). Annexin II was used as a marker and loading control for membrane proteins. To detect secreted GRP78/BiP, RD cells were maintained in DMEM without FBS and infected with EV71 2231 (MOI of 10) for 1 h. The cells were then washed with PBS and cultured in DMEM supplemented with 2% FBS for 2 h, followed by culturing in DMEM without FBS for another 6 h. The cell culture medium and cell lysates were collected as described in the secretion assay above. The secretion of GRP78/BiP did not differ between mock- and virus-infected cells (lanes 3 and 4, ), although GRP78/BiP was upregulated by EV71 (lanes 1 and 2, ). The same results were obtained in virus-infected HEK293T cells and SF268 cells (a human neuroblastoma cell line) (data not shown). In addition, previous studies have shown that GRP78/BiP devoid of C-terminal KDEL motif is secreted.Citation31 Using the cytosolic/microsomal fractionation assay, we detected both the KDEL recognition (motif) and GRP78/BiP in the cytosolic fraction (). These results further confirmed that EV71 infection did not trigger GRP78/BiP secretion. We concluded that EV71 virus infection induces the intracellular redistribution of GRP78/BiP from the ER to cytoplasm.
Figure 1 Subcellular fractionation profiles of GRP78/BiP in mock-infected, EV71-infected or DTT-treated RD cells. The PNSs from mock-infected, EV71-infected (MOI of 10, 6 h p.i.), 2.5 mM DTT-treated (6 h) cells were fractionated using OptiPrep gradients. The fractions were collected from the top and subjected to SDS–PAGE and subsequent western blotting using specific antibodies against GRP78/BiP, calnexin, and viral proteins 3A and 2C. The results are representative of three independent experiments.
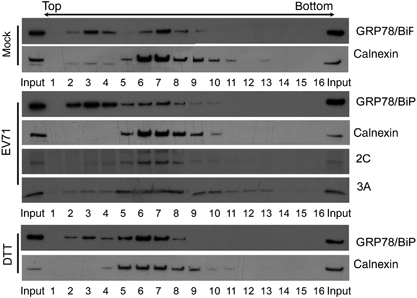
Figure 2 EV71 infection induced an intracellular redistribution of GRP78/BiP. (A, B and E) Cytoplasmic/microsomal localization of GRP78/BiP during viral infection. Homogenates extracted from mock, EV71-infected (MOI of 10, 6 h p.i.) or DTT-treated (2.5 mM, 6 h) RD (A and E) or HEK293T cells (B) were subjected to cytosolic/microsomal fractionations. After centrifugation, fractionates were resolved on SDS–PAGE followed by western blotting with antibodies against the indicated proteins. (C) Cell surface distribution of GRP78/BiP in EV71-infected RD cells. RD cells were infected with EV71 at an MOI of 10, and the membrane proteins were prepared using a Plasma Membrane Protein Extraction Kit at 6 h p.i. Annexin II and GAPDH served as markers for the plasma membrane and cytosol fractions, respectively. (D) Immunoblot analysis of cell lysates and culture medium obtained from mock-infected or EV71-infected cultures. Thirty micrograms of lysates or Amicon filter unit-concentrated supernatant was subjected to SDS–PAGE and western blotting. Calnexin, GAPDH and β-actin were used as markers for microsomes, cytoplasm and internal controls, respectively. The results are representative of three independent experiments. An asterisk marks nonspecific bands.
Cytoplasmic elevation of GRP78/BiP does not result from virus-induced KDEL receptor diminution
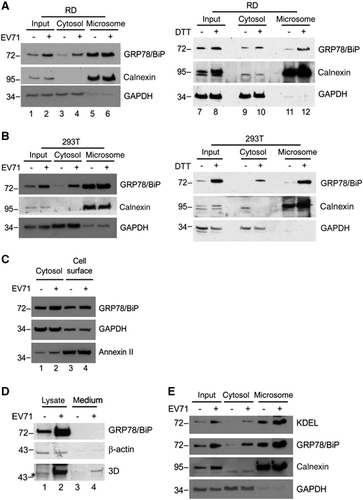
The KDEL receptor (KDELR) is primarily located in the Golgi complex and the ER-Golgi intermediate compartment. It recognizes and retrieves KDEL-containing proteins from the Golgi complex to transport them to the ER.Citation31, Citation32 Thus, we examined the effect of EV71 infection on KDELR expression and its association with the redistribution of GRP78/BiP. We infected RD cells with EV71 and collected cell lysates at the indicated time points p.i. to monitor the expression level of KDELR. The 3D intensity of viral protein became detectable starting at 4 h p.i. and increased in a time-dependent manner until 6 h p.i. However, decreased KDELR levels were detected in EV71-infected cells relative to mock-infected controls after 5 h p.i. (). We further examined the role of KDELR in virus-induced GRP78/BiP redistribution using cytosolic/microsomal fractionation assay in cells overexpressing KDELR. KDELR tagged with HA (KDELR-HA) was transfected into RD cells, and its expression levels were similar in mock- and virus-infected cells, as monitored by western blotting (lanes 3 and 4 of bottom panel, ). However, the overexpression of KDELR did not affect virus-induced GRP78/BiP redistribution into the cytosol (lanes 5–12, ), indicating that the virus-induced KDELR decreases are not associated with the redistribution of GRP78/BiP. This finding also suggests that the reduction of KDELR upon viral infection might be a result of virus-induced translational attenuation.
Figure 3 Redistribution of GRP78/BiP was independent of the expression of KDEL receptor. (A) RD cells were infected with EV71 (MOI of 10), followed by whole-cell lysate preparation for western blot analysis of the expression levels of KDELR. The viral protein expression is indicated by the levels of viral protein 3D. (B) RD cells were transfected with KDELR-HA or vector control for 24 h and then infected with EV71 (MOI of 10). Cell homogenates were collected at 6 h p.i. and subjected to cytosolic/microsomal fractionations and western blot analysis for the distribution of GRP78/BiP. The results are representative of three independent experiments. The expression of KDELR-HA was detected with anti-HA antibodies (bottom panel).
dsRNA is required for the upregulation and redistribution of GRP78/BiP
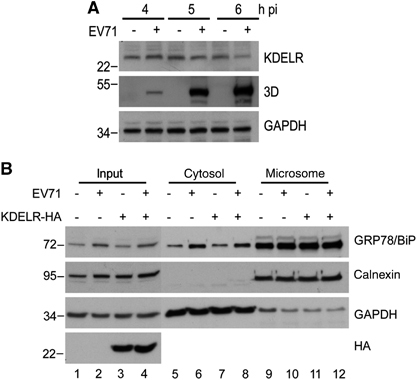
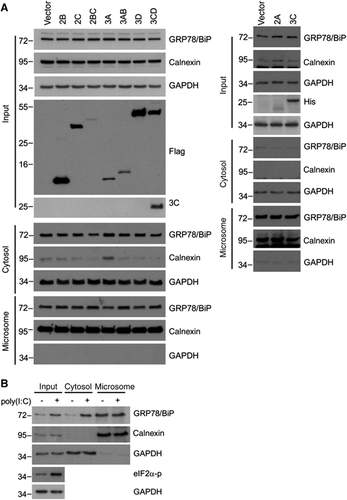
We previously showed that the EV71-induced UPR depended on viral replication in host cells because replication-incompetent EV71 due to UV-inactivation no longer exhibited UPR-induction activity.Citation23 To explore the necessity of viral replication for GRP78/BiP redistribution, we infected RD cells with mock- or UV-irradiated EV71 (UV-EV71), and subjected the resultant cell homogenates to subcellular fractionation. In contrast to mock-treated EV71, UV-EV71 did not increase the cytosolic GRP78/BiP level (lanes 5 and 6, ), indicating that viral replication is necessary for GRP78/BiP redistribution. Next, we explored the ability of viral proteins or replicative viral RNA to upregulate GRP78/BiP and increase its cytoplasmic accumulation. HEK293T cells transfected with individual nonstructural proteins or with poly(I:C), a mimic viral replication intermediate,Citation33 were lysed, and the lysates were subjected to western blotting. The results showed dsRNA induced not only the phosphorylation of its downstream target PKR but also resulted in substantial GRP78/BiP expression (). Conversely, the expression of individual viral proteins did not result in elevated levels of GRP78/BiP, although the viral proteins were efficiently expressed (). The quantitative PCR data showed that transfection with poly(I:C) did not increase the mRNA level of GRP78/BiP (), which corroborated the effect of EV71 infectionCitation23 and indicated that GRP78/BiP is posttranscriptionally regulated regulation by dsRNA. We further determined whether the cytosolic accumulation of GRP78/BiP in response to virus infection was due to dsRNA or the expression of viral proteins. No individual protein changed the expression level of cytosolic GRP78/BiP (), and only in poly(I:C)-transfected cells exhibited increases in cytosolic GRP78/BiP (), indicating a role for dsRNA in the redistribution of GRP78/BiP. Therefore, dsRNA, not the viral protein, is responsible for GRP78/BiP upregulation and cytosolic redistribution.
Figure 4 dsRNA, but not viral proteins, induced the upregulation of GRP78/BiP. (A) Immunoblot analysis of GRP78/BiP in cytosolic/microsomal fractionations from mock-infected, EV-infected (MOI of 10, 6 h p.i.) or UV-EV71-infected RD cells. The results are representative of three independent experiments. (B and C) HEK293T cells were transfected with 10 μg/mlof dsRNA analogue poly(I:C) for 18 h (B) or Flag or His fusion constructs of individual viral proteins for 24 h (C). The success of poly(I:C) transfection was assessed based on the phosphorylation of PKR (B). Whole-cell lysates were analyzed for the viral protein and GRP78/BiP expression levels by western blotting. The expression levels of individual viral proteins were monitored with anti-Flag or anti-His antibodies as indicated (C). The results are representative of three independent experiments. (D) Expression level of GRP78/BiP mRNA in poly(I:C) transfection. RD cells were transfected with poly(I:C) for 18 h, and the GRP78/BiP mRNA was detected using quantitative real-time RT-PCR. The results are expressed as the means±s.d. (n=3). The difference was not significant.
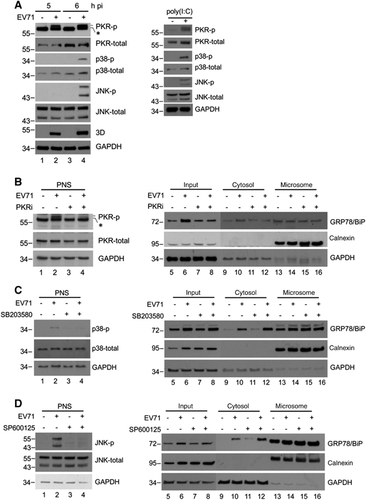
PKR mediates virus-induced elevation of cytosolic GRP78/BiP
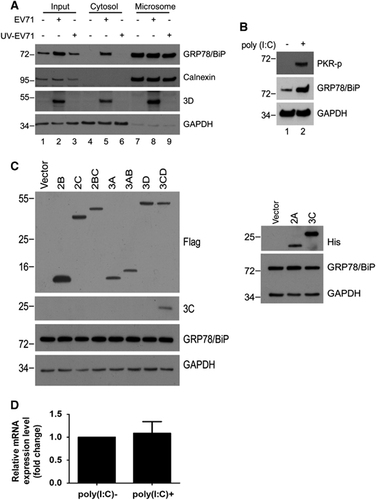
Previous reports indicated that PKR, p38 MAPK, and JNK may be activated by dsRNA or virus infection.Citation34, Citation35, Citation36 Therefore, we confirmed the phosphorylation of PKR, p38 MAPK and JNK in poly(I:C)-treated or virus-infected RD cells (). The results showed that the activation of all these kinases was detectable in both virus-infected and poly(I:C)-treated cells, and their expression levels were similar to those of the mock-infected controls (), indicating that the changes in active kinase levels were not caused by an increase in the expression of total kinases. We next attempted to identify the kinase affects the cytosolic redistribution of GRP78/BiP. We treated cells with inhibitors of PKR (PKRi), p38 MAPK (SB203580) and JNK (SP600125) after virus infection, and the resultant cell homogenates were subjected to subcellular fractionation (). The inhibitors effectively abolished the virus-induced phosphorylation in the PNS (lanes 1–4, ). It is possible that P38 MAPK and JNK did not cause the redistribution because the levels of virus-induced cytosolic GRP78/BiP in SB203580- or SP600125-treated cells were similar to those in control DMSO-treated cells (lanes 6 and 8, ). However, virus-induced cytosolic GRP78/BiP increases were not observed in cells treated with PKRi (lanes 6 and 8, ), indicating that the inhibition of PKR, but not p38 MAPK or JNK, impaired the accumulation of cytosolic GRP78/BiP in response to EV71 infection.
Figure 6 dsRNA induced a PKR-dependent cytosolic elevation of GRP78/BiP on EV71 infection. (A) EV71 infection and poly(I:C) transfection led to the phosphorylation of PKR, p38 MAPK and JNK. RD cells were infected with EV71 (MOI of 10, 5 and 6 h, respectively) or transfected with poly(I:C) for 18 h. Cells were lysed for western blotting with antibodies against the indicated proteins. Antibodies against total PKR, total p38 MAPK, total JNK and GAPDH were used to monitor loading. An asterisk marks a nonspecific band. (B–D) RD cells were infected with EV71 (MOI of 10) or mock-infected and subsequently treated with DMSO, SP600125 (20 μM), SB203580 (20 μM) or PKRi (10 μM) at 3 h p.i. The PNS was collected at 6 h p.i. to examine the levels of p-JNK, p-p38 MAPK and p-PKR by western blotting (left) or subjected to cytosolic/microsomal fractionations to analyze the distribution of GRP78/BiP with a western blot (right). An asterisk marks nonspecific bands. These results are representative of three independent experiments.
ISR and apoptosis does not cause the upregulation of cytosolic GRP78/BiP
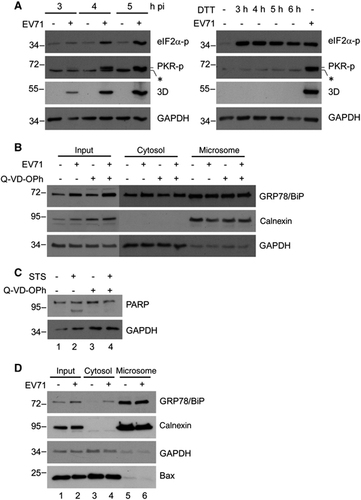
Notably, although both virus infection and DTT treatment induced eIF2α phosphorylation (), DTT treatment did not increase the level of cytosolic GRP78/BiP (), ruling out the involvement of ISR in the redistribution of GRP78/BiP. In addition, PKR phosphorylation was observed in cells infected with EV71, but not in cells treated with DTT (), supporting that virus-induced PKR phosphorylation plays a role in GRP78/BiP modulation. Together, these results suggest that PKR is required for the virus-induced increase in cytosolic GRP78/BiP. Because recent studies have shown that PKR mediates apoptosis,Citation37 we examined whether the accumulation of cytosolic GRP78/BiP occurs concomitantly with virus-induced apoptosis. To this end, DMSO or Q-VD-OPh, a broad caspase inhibitor, was added to EV71-infected and uninfected cells, and the resultant cell homogenates were subjected to subcellular fractionation. Treatment with Q-VD-OPh did not affect the EV71-induced accumulation of cytosolic GRP78/BiP (). Because caspase activity was difficult to detect by a western blot analysis 6 h p.i., the inhibition efficiency of apoptosis by Q-VD-OPh was demonstrated in a parallel experiment, in which staurosporine-induced poly-(ADP-ribose) polymerase (PARP) cleavage was reduced in response to Q-VD-OPh treatment (). Furthermore, because the ER stress-induced translocation of Bax protein to the ER membrane has been shown to be critical for the release of ER luminal proteins by increasing membrane permeability during ER stress-induced apoptosis,Citation38, Citation39 we next investigated the localization of Bax protein upon EV71 infection. The results of the cytosolic/microsomal fractionation assays showed that virus infection did not alter the subcellular localization of Bax (lanes 5 and 6 of bottom panel, ). Together, these results revealed that the upregulation of cytosolic GRP78/BiP is not apparently related to virus (PKR)-induced apoptosis.
Figure 7 EV71-induced cytosolic elevation of GRP78/BiP did not occur via ISR or apoptosis. (A) Time-course study of p-PKR and p-eIF2α in EV71-infected (MOI of 10, left) or DTT-treated (2.5 mM, right) RD cells. An asterisk marks a nonspecific band. (B) RD cells were infected with or without EV71 (MOI of 10) and subsequently treated with DMSO or Q-VD-OPh (20 μM) at 0 h p.i. Cell homogenates were collected at 6 h p.i. and subjected to cytosolic/microsomal fractionations and a western blot analysis of the distribution of GRP78/BiP. (C) Inhibition of staurosporine-induced PARP-cleavage by Q-VD-OPh. RD cells were treated with 100 nM of staurosporine (STS) in the presence or absence of Q-VD-OPh (20 μM) for 4 h. The cell lysates were collected and subjected to a western blot analysis. (D) Relocalization of Bax during viral infection. Cell homogenates extracted from mock- or EV71-infected (MOI of 10, 6 h p.i.) RD cells were subjected to cytosolic/microsomal fractionations. After centrifugation, fractionates were resolved on the SDS–PAGE followed by western blotting with antibodies against the indicated proteins. The results are representative of three independent experiments.
Sustained ER stress caused by the EV71-induced redistribution of GRP78/BiP is advantageous to the virus
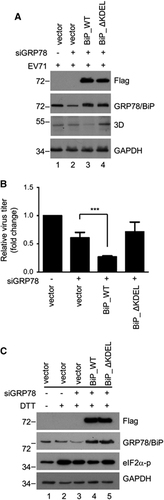
To explore the effect of GRP78/BiP redistribution on viral protein expression and viral particle production, siRNA-resistant wild-type (GRP78/BiP_WT) or C-terminal KDEL-deleted mutant GRP78/BiP (GRP78/BiP_ΔKDEL) devoid of the ER retention signal KDEL was expressed in RD cells in which endogenous GRP78/BiP was knocked down with specific siRNA. After infection with EV71, the expression level of viral protein and the virus titer were assessed by western blotting and a plaque assay, respectively. Both GRP78/BiP_WT and GRP78/BiP_ΔKDEL were effectively and equally expressed in endogenous GRP78/BiP-silenced RD cells (lanes 3 and 4, ). Compared with the vector control, the re-expression of GRP78/BiP_WT suppressed viral protein expression, as indicated by viral 3D protein and viral particle formation, but viral protein expression was not significantly affected in GRP78/BiP_ΔKDEL re-expressing cells (). Because ER stress is beneficial for virus replication,Citation23 we hypothesized that GRP78/BiP was arrested in the cytosol, leading to sustained ER stress upon virus infection. To verify this hypothesis, the level of DTT-induced eIF2α phosphorylation in GRP78/BiP_WT or GRP78/BiP_ΔKDEL re-expressing cells was investigated. As expected, the DTT-induced eIF2α phosphorylation level decreased in response to transfection with GRP78/BiP_WT, but this phosphorylation was not affected in GRP78/BiP_ΔKDEL re-expressing cells (lanes 4 and 5, ). Taken together, these results indicate that EV71 infection induces permanent ER stress by redistributing GRP78/BiP to facilitate virus infection.
Figure 8 EV71-induced redistribution of GRP78/BiP conferred prolonged ER stress and was advantageous to virus infection. (A–C) RNAi rescue experiments were performed by transfecting GRP78/BiP-knockdown RD cells with siRNA-resistant BiP_WT or BiP_ΔKDEL. The cells were then infected with EV71 (MOI of 10, 6 h p.i.). The viral protein expression, as indicated by the 3D level, was assessed by a western blot analysis (A), and the virus titers were determined by plaque assays (B). The plaque assay results are expressed as the means±s.d. (n=3). ***P<0.001 compared with the re-expression of vector alone. (C) The cells were treated with 2.5 mM DTT for 4 h, and the cell lysates were then collected to examine the levels of p-eIF2α with a western blotting analysis. These results are representative of three independent experiments.
DISCUSSION
The ER is a crucial organelle that supports picornavirus replication. It is considered a source of precursor membranes for viral replication complex formation.Citation40, Citation41, Citation42 In addition, our previous study showed that viral 2C protein interacts with the host ER transmembrane protein reticulon 3 and is required for EV71 replication.Citation26 Recently, the ER-derived autophagosome-like vesicle that participates in nonlytic viral spread and virus infection has emerged as a topic in picornavirus studies.Citation43, Citation44 Therefore, learning how viruses modulate the ER membrane or ER proteins may provide new insights for the antiviral research.
We previously demonstrated that EV71 infection increased the GRP78/BiP protein levels in RD cells.Citation23 Paradoxically, the ectopic expression of GRP78/BiP not only alleviated ER stress but also inhibited viral replication. In this study, results from the OptiPrep gradient showed that EV71 infection altered the distribution of GRP78/BiP (). We further performed secretory assays, plasma membrane protein extraction assays, and cytosolic/microsomal fractionation assays to show that EV71 infection increased the level GRP78/BiP in the cytosol but not in the ER (). We suggest that this cytosolic increase in GRP78/BiP facilitates viral replication.
The expression of GRP78/BiP is conventionally regulated at both the transcriptional and posttranscriptional levels. The GRP78/BiP promoter is primarily activated by ER stress-response elements (ERSEs) under stressed conditions, and these elements share a consensus sequence: CCAAT(N9)CCACG.Citation45 In addition, the ATF4-binding site has also been reported to localize to an ATF/CRE sequence upstream of the ERSEs and contribute to GRP78/BiP induction.Citation46 The posttranscriptional regulation of GRP78/BiP is mediated by the activation of internal ribosome entry sequence (IRES) in the 5′ untranslated region of GRP78/BiP mRNACitation47 or by an increase in protein stability.Citation48, Citation49
Some viruses enforce the expression of GRP78/BiP. For example, human cytomegalovirus induces GRP78/BiP by increasing transcription and translation.Citation50 Hepatitis C virus E2 envelope protein activates the GRP78/BiP promoter.Citation51 We previously reported that EV71 infection induces GRP78/BiP in an ATF6-independent manner, but this induction does not depend on the activation of ERSE promoter of GRP78/BiP.Citation23 The quantitative PCR results demonstrated that dsRNA posttranscriptionally upregulates GRP78/BiP expression (). We postulate that EV71 infection activates GRP78/BiP IRES and leads to GRP78/BiP accumulation. In support of this hypothesis, previous studies showed that dsRNA guides translational repression,Citation52, Citation53 and IRES-mediated translation is enhanced when cap-dependent protein synthesis is decreased.Citation54 Furthermore, poliovirus, a member of the Picornaviridae family, reportedly increases the translation of GRP78/BiP mRNA in a cap-independent manner.Citation55 PI3K/Akt has also been shown to promote GRP78/BiP stability under conditions of ER stress.Citation48 Because EV71 infection induces PI3K/Akt activation,Citation25 we cannot exclude the possibility that EV71 might enhance GRP78/BiP stability by activating PI3K/Akt. In addition, as posttranscriptional regulators, microRNAs also play a role in GRP78/BiP regulation. miR-181 has been reported to directly target GRP78/BiP mRNA and repress its expression by suppressing translation. In our current study, we found that EV71 infection leads to a decrease in miR-181 (unpublished data). Thus, the mechanism by which EV71 infection upregulates GRP78/BiP expression will be an interesting topic for future studies.
Some mechanisms have been reported for the redistribution of GRP78/BiP to the cytosol: (1) GRP78/BiP is relocated to the cytoplasm via ER-associated degradation (ERAD);Citation16 (2) Bcl-2 family proteins, including Bax and Bak, modulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis;Citation38 and (3) cytosolic GRP78va that lacks the ER signaling peptide is generated by the alternative splicing of mRNA.Citation14 In our previous reports, we demonstrated that EV71 upregulated the XBP1 mRNA level, but the IRE1-mediated XBP1 splicing and transcription of genes that encode ERAD associated proteins was not activated,Citation23, Citation28 indicating that EV71 infection does not activate ERAD. In this study, we found that virus infection did not change the electrophoretic mobility of the GRP78/BiP and the subcellular localization of Bax (), implying a not-yet-identified mechanism of GRP78/BiP regulation upon EV71 infection. Interestingly, the data obtained from cells transfected with dsRNA or individual viral protein indicated that only dsRNA was involved in the cytosolic elevation of GRP78/BiP (). Thus, the dsRNA-mediated signaling pathway was hypothesized to cause the redistribution of GRP78/BiP. We examined three dsRNA signaling pathways, and only the inhibition of PKR activity significantly attenuated the virus-induced upregulation of cytosolic GRP78/BiP (). Therefore, we suggest that the dsRNA-PKR signaling axis is involved in the EV71-induced redistribution of GRP78/BiP.
Figure 5 dsRNA, but not viral proteins, is responsible for the redistribution of GRP78/BiP. Immunoblot analysis of GRP78/BiP in cytosolic and microsomal fractions from cells transfected with viral proteins (A) or poly(I:C) (B). These results are representative of three independent experiments.
The investigation of the effect of GRP78/BiP redistribution on viral protein expression and viral particle production indicates that permanent ER stress caused by the redistribution of GRP78/BiP is beneficial to EV71 (), which is consistent with our previous study.Citation23 However, because other roles have been reported for cytosolic GRP78/BiP, GRP78/BiP redistribution may facilitate EV71 replication in many ways. GRP78/BiP reportedly protects cells against ER stress-induced apoptosis by forming a complex with caspase-7 and caspase-12 to prevent the release of caspase-12 from the ER.Citation56 In addition, GRP78va can interact with P58IPK and reduce its protein level, which in turn specifically enhances PERK signaling and protects cells from ER stress-induced cell death.Citation14 Accordingly, growing evidence shows that viruses may suppress apoptosis for the efficient production of mature virus, which maximizes viral infectivity;Citation57, Citation58 therefore, the cytosolic relocalization of GRP78/BiP may promote viral infectivity by suppressing apoptosis. Furthermore, an interaction between HBV precore protein and cytosolic GRP78/BiP may enhance viral persistence.Citation16 In our study, viral RNA-dependent RNA polymerase 3D cofractionated with GRP78/BiP both in the lighter fractions of the OptiPrep gradient (data not shown) and the cytosolic part of cytosolic/microsomal fractionation () upon EV71 infection. Thus, cytosolic GRP78/BiP may play a role in facilitating viral protein folding or enhancing virus replication. In summary, our findings suggest that PKR-mediated GRP78/BiP redistribution is proviral for EV71. This should be further explored as a potential antiviral target for the inhibition of EV71.
We thank Drs Shin-Ru Shih (Research Center for Emerging Viral Infections, Chang Gung University, Taoyuan, Taiwan), Mei-Shang Ho (Institute of Biomedical Sciences, Academia Sinica, Nankang, Taiwan) and Guanghui Wang (Laboratory of Molecular Neuropathology, Chinese Academy of Sciences, Hefei, Anhui, China) for the reagents. This work was supported by grants from Chang Gung Memorial Hospital (CMRPD1B0423, CMRPD1D0322 and CMRPD1A0163) and the Ministry of Science and Technology of Taiwan (MOST103-2325-B-182-009 and 101-2321-B-001-013).
Related Research Data
References
- RacanielloVR.Picornaviridae: The viruses and their replication. In: David M, Knipe PMH, Malcolm A Martin, Diane E Griffin, Robert A Lamb, Bernard Roizman, Stephen E Straus(eds).Fields Virology,5th ednvol. 1.Lippincott Williams & Wilkins: Philadelphia.2007,795–838.
- SolomonT,LewthwaiteP,PereraDet al.Virology, epidemiology, pathogenesis, and control of enterovirus 71.Lancet Infect Dis2010; 10:778–790.
- FengQ,HatoSV,LangereisMAet al.MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells.Cell Rep2012; 2:1187–1196.
- NanduriS,CarpickBW,YangYet al.Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation.EMBO J1998; 17:5458–5465.
- LiuL,WiseDR,DiehlJAet al.Hypoxic reactive oxygen species regulate the integrated stress response and cell survival.J Biol Chem2008; 283:31153–31162.
- KozutsumiY,NormingtonK,PressEet al.Identification of immunoglobulin heavy chain binding protein as glucose-regulated protein 78 on the basis of amino acid sequence, immunological cross-reactivity, and functional activity.J Cell Sci Suppl1989; 11:115–137.
- FlahertyKM,DeLuca-FlahertyC,McKayDB.Three-dimensional structure of the ATPase fragment of a 70 K heat-shock cognate protein.Nature1990; 346:623–628.
- ZhuX,ZhaoX,BurkholderWFet al.Structural analysis of substrate binding by the molecular chaperone DnaK.Science1996; 272:1606–1614.
- WangH,KurochkinAV,PangYet al.NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: a preview of chaperone-protein interaction.Biochemistry1998; 37:7929–7940.
- FreemanBC,MyersMP,SchumacherRet al.Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1.EMBO J1995; 14:2281–2292.
- NiM,ZhangY,LeeAS.Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting.Biochem J2011; 434:181–188.
- ZhangY,TsengCC,TsaiYLet al.Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production.PLoS One2013; 8:e80071.
- MisraUK,Gonzalez-GronowM,GawdiGet al.A novel receptor function for the heat shock protein Grp78: silencing of Grp78 gene expression attenuates alpha2M*-induced signalling.Cell Signal2004; 16:929–938.
- NiM,ZhouH,WeySet al.Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP.PLoS One2009; 4:e6868.
- JindadamrongwechS,ThepparitC,SmithDR.Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2.Arch Virol2004; 149:915–927.
- DuriezM,RossignolJM,SitterlinD.The hepatitis B virus precore protein is retrotransported from endoplasmic reticulum (ER) to cytosol through the ER-associated degradation pathway.J Biol Chem2008; 283:32352–32360.
- LeeAS.The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.Methods2005; 35:373–381.
- JhengJR,HoJY,HorngJT.ER stress, autophagy, and RNA viruses.Front Microbiol2014; 5:388.
- ChanSW.Unfolded protein response in hepatitis C virus infection.Front Microbiol2014; 5:233.
- KePY,ChenSS.Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro.J Clin Invest2011; 121:37–56.
- PenaJ,HarrisE.Dengue virus modulates the unfolded protein response in a time-dependent manner.J Biol Chem2011; 286:14226–14236.
- SuHL,LiaoCL,LinYL.Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response.J Virol2002; 76:4162–4171.
- JhengJR,LauKS,TangWFet al.Endoplasmic reticulum stress is induced and modulated by enterovirus 71.Cell Microbiol2010; 12:796–813.
- ChangCW,LeuYL,HorngJT.Daphne Genkwa sieb. Et zucc. Water-soluble extracts act on enterovirus 71 by inhibiting viral entry.Viruses2012; 4:539–556.
- WongWR,ChenYY,YangSMet al.Phosphorylation of PI3K/Akt and MAPK/ERK in an early entry step of enterovirus 71.Life Sci2005; 78:82–90.
- TangWF,YangSY,WuBWet al.Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication.J Biol Chem2007; 282:5888–5898.
- YamayoshiS,YamashitaY,LiJet al.Scavenger receptor B2 is a cellular receptor for enterovirus 71.Nat Med2009; 15:798–801.
- JhengJR,LinCY,HorngJTet al.Inhibition of enterovirus 71 entry by transcription factor XBP1.Biochem Biophys Res Commun2012; 420:882–887.
- WangP,LiB,ZhouLet al.The KDEL receptor induces autophagy to promote the clearance of neurodegenerative disease-related proteins.Neuroscience2011; 190:43–55.
- WuYP,ChangCM,HungCYet al.Japanese encephalitis virus co-opts the ER-stress response protein GRP78 for viral infectivity.Virol J2011; 8:128.
- MunroS,PelhamHR.A C-terminal signal prevents secretion of luminal ER proteins.Cell1987; 48:899–907.
- CancinoJ,CapalboA,Di CampliAet al.Control systems of membrane transport at the interface between the endoplasmic reticulum and the Golgi.Dev Cell2014; 30:280–294.
- AlexopoulouL,HoltAC,MedzhitovRet al.Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3.Nature2001; 413:732–738.
- LemairePA,AndersonE,LaryJet al.Mechanism of PKR Activation by dsRNA.J Mol Biol2008; 381:351–360.
- PengH,ShiM,ZhangLet al.Activation of JNK1/2 and p38 MAPK signaling pathways promotes enterovirus 71 infection in immature dendritic cells.BMC Microbiol2014; 14:147.
- PetersKL,SmithHL,StarkGRet al.IRF-3-dependent, NFkappa B- and JNK-independent activation of the 561 and IFN-beta genes in response to double-stranded RNA.Proc Natl Acad Sci USA2002; 99:6322–6327.
- GilJ,EstebanM.Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action.Apoptosis2000; 5:107–114.
- WangX,OlberdingKE,WhiteCet al.Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis.Cell Death Differ2011; 18:38–47.
- ZongWX,LiC,HatzivassiliouGet al.Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis.J Cell Biol2003; 162:59–69.
- TraheyM,OhHS,CameronCE,HayJC.Poliovirus infection transiently increases COPII vesicle budding.J Virol2012; 86:9675–9682.
- SchlegelA,GiddingsTHJr,LadinskyMSet al.Cellular origin and ultrastructure of membranes induced during poliovirus infection.J Virol1996; 70:6576–6588.
- van KuppeveldFJ,HoenderopJG,SmeetsRLet al.Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release.EMBO J1997; 16:3519–3532.
- ChenYH,DuW,HagemeijerMCet al.Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses.Cell2015; 160:619–630.
- BirdSW,MaynardND,CovertMWet al.Nonlytic viral spread enhanced by autophagy components.Proc Natl Acad Sci USA2014; 111:13081–13086.
- LiWW,AlexandreS,CaoXet al.Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin.J Biol Chem1993; 268:12003–12009.
- LuoS,BaumeisterP,YangSet al.Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements.J Biol Chem2003; 278:37375–37385.
- MacejakDG,SarnowP.Internal initiation of translation mediated by the 5' leader of a cellular mRNA.Nature1991; 353:90–94.
- DaiRY,ChenSK,YanDMet al.PI3K/Akt promotes GRP78 accumulation and inhibits endoplasmic reticulum stress-induced apoptosis in HEK293 cells.Folia Biol (Praha)2010; 56:37–46.
- OuyangYB,LuY,YueSet al.miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo.Neurobiol Dis2012; 45:555–563.
- BuchkovichNJ,YuY,PiercieyFJJret al.Human cytomegalovirus induces the endoplasmic reticulum chaperone BiP through increased transcription and activation of translation by using the BiP internal ribosome entry site.J Virol2010; 84:11479–11486.
- LibermanE,FongYL,SelbyMJet al.Activation of the grp78 and grp94 promoters by hepatitis C virus E2 envelope protein.J Virol1999; 73:3718–3722.
- PriceNT,WelshGI,ProudCG.Phosphorylation of only serine-51 in protein synthesis initiation factor-2 is associated with inhibition of peptide-chain initiation in reticulocyte lysates.Biochem Biophys Res Commun1991; 176:993–999.
- PestovaTV,de BreyneS,PisarevAVet al.eIF2-dependent and eIF2-independent modes of initiation on the CSFV IRES: a common role of domain II.EMBO J2008; 27:1060–1072.
- GerlitzG,JagusR,Elroy-SteinO.Phosphorylation of initiation factor-2 alpha is required for activation of internal translation initiation during cell differentiation.Eur J Biochem2002; 269:2810–2819.
- SarnowP.Translation of glucose-regulated protein 78/immunoglobulin heavy-chain binding protein mRNA is increased in poliovirus-infected cells at a time when cap-dependent translation of cellular mRNAs is inhibited.Proc Natl Acad Sci USA1989; 86:5795–5799.
- RaoRV,PeelA,LogvinovaAet al.Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78.FEBS Lett2002; 514:122–128.
- CallusBA,VauxDL.Caspase inhibitors: viral, cellular and chemical.Cell Death Differ2007; 14:73–78.
- UrbanowskiMD,HobmanTC.The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism.J Virol2013; 87:872–881.