
Abstract
The Group B streptococcus (GBS) can express a capsular polysaccharide (CPS). There are ten recognized CPSs (Ia, Ib, and II–IX). A GBS isolate is considered nontypeable (NT) when CPS cannot be identified as one of ten types. Two groups of GBS NT isolates were studied, isolates without surface sialic acid (sia(−)) and isolates with surface sialic acid (sia(+)). The first objective was to characterize NT sia(−) isolates that failed CPS identification by an immunodiffusion antisera typing assay and a RT-PCR capsule typing assay. NT sia(−) isolates were characterized by assaying phenotypic changes and identifying covR/S mutations that may potentially have a role in the altered phenotypes. The second objective was to characterize NT sia(+) isolates that failed to identify as one of the ten CPS types by an immundiffusion antisera-based typing assay and a RT-PCR capsule typing assay yet expressed capsule. Fifteen NT sia(−) isolates displayed increased β hemolysis/orange pigmentation, decreased CAMP activity, inability to form biofilm, and susceptibility to phagocytosis by human blood. DNA sequence analysis of the covR/S genes in the sia(−) isolates found mutations in 14 of 15 isolates assayed. These mutations in the covR/S genes may potentially contribute to lack of expression of phenotypic traits assayed in vitro. For the three NT sia(+) isolates, whole-genome sequence analyses identified two isolates with cps gene clusters identical to the recently described and uncommon CPSIIa type. The third isolate possessed a hybrid cluster containing cps genes for both CPSIIa and CPSV suggesting recombination between these two gene clusters.
Introduction
Group B streptococci, GBS, (also referred as Streptococcus agalactiae) are a leading cause of invasive infections which can manifest as pneumonia, septicemia, and meningitis in neonates and a serious cause of morbidity and/or mortality in adults with underlying diseasesCitation1–Citation7. GBS produce an array of virulence factors which include a β-hemolysin/cytolysin which can cause a narrow zone of β-hemolysis on 5% sheep blood agar, a CAMP factor that lyses sheep red blood cells previously sensitized with a sphingomyelinase produced by some Staphylococcus aureus strainsCitation8,Citation9, and a capsular polysaccharide (CPS) of which there are ten types (Ia, Ib, and II–IX)Citation10–Citation13. These ten types of CPS can either be identified serologically or by various molecular assays. Those GBS that cannot be CPS typed are termed nontypable (NT). The NT phenotype of some GBS isolates may be due to the expression of undetectable amount of CPS by serological methods, lack of CPS expression, or production of uncharacterized capsular polysaccharide for which CPS typing antibodies not yet are availableCitation14.
Functions of CPS include protection of GBS from being killed by host immune cells such as macrophages and is a key component in the process of biofilm formation in the presence of human plasmaCitation15. Inactivation of CPS biosynthesis gene(s) reduces resistance toward phagocytic killingCitation16 and inhibition of biofilm formationCitation15.
GBS CPS of the ten CPS variants are formed by different arrangements of four component sugars (glucose, galactose, N-acetylglucosamine, and sialic acid) into a unique repeating unitCitation17–Citation20. All recognized GBS CPS types have sialic acid attached to their CPS structureCitation17–Citation20 that can interfere with complement-mediated killing by host immune cellsCitation21,Citation22. Based on the conservation of sialic acid among all recognized GBS CPS types and the essentiality of its presence for full capsule biosynthesis and expressionCitation17,Citation18, sialic acid has been utilized as recognition marker for GBS CPS productionCitation23.
Expression of GBS virulence factors, such as CPS, is controlled by the two-component system CovR/S. CovR/S is a major global regulatory system that is responsible for modulating the transcription of up to 7% of total GBS genomic genesCitation24 including GBS virulence genes such β-h/cCitation25-Citation29, CAMP factorCitation25, cell surface proteinsCitation25, capsule, and surface sialic acid expressionCitation30. Mutations in covR/S have been shown to alter the phenotypic expression of these virulence factorsCitation24.
We reported previously that 9% of GBS isolates in Alberta, Canada, collected from patients with invasive diseases between 2003 and 2013 were identified as NTCitation31 and of those, 52.8% were sialic acid negative (sia(−))Citation23. The sia(−) phenotype in NT GBS isolates suggests loss of cps expression as sialic acid is present on all known GBS CPS types. This suggested that the NT GBS isolates that were sia(−) in our collection may have genetic mutations in the covR/S genes resulting in a loss of CPS expressionCitation25,Citation27,Citation28. We also previously identified from this collection three sialic positive (sia(+)) GBS isolates that could not be CPS typed by a serological assay or by PCRCitation23.
The first objective was to characterize NT sia(−) isolates that failed CPS identification by an immunodiffusion antisera-based typing assay and a RT-PCR capsule typing assay. The second objective was to characterize NT sia(+) isolates that failed to identify as one of the ten CPS types by an immundiffusion antisera-based typing assay and a RT-PCR capsule typing assay yet expressed capsule. This characterization included the identification of potential new cps gene clusters among these isolates using whole-genome sequencing.
Results
Sia(−) isolates
Phenotypic differences between GBS NT sia(−) isolates and GBS wild-type strain COHI
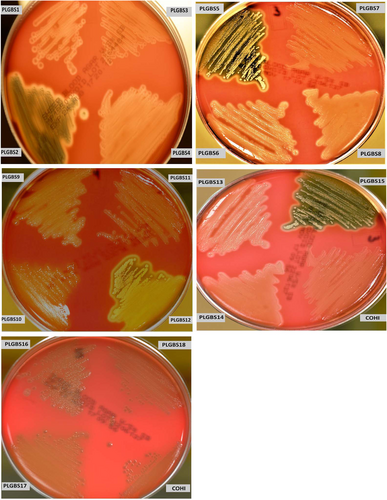
Analysis of β-hemolytic activity in our collection of sia(−) isolates found that 15 sia(−) NT isolates (18%) exhibited increased β-hemolytic activity on 5% sheep blood agar compared to the positive control GBS isolate COH1 (Fig. ). These were designated PLGBS1–15. The increase in the hemolytic activity on sheep blood agar plates was confirmed by a microtitre hemolysis assay, which showed an average fold increase of 3.5× (±0.8) (ranged from 2.6 to 4.36) over the control GBS strain COHI (Table ). In addition to the increased hemolytic activity, these 15 sia(−) isolates visually displayed greater orange pigmentation vs. control (GBS strain COHI) as shown in culture pellets (supplemental Figure 1). This is expected as β-hemolysis and orange pigment production are caused by the same toxin, an ornithine rhamnolipidCitation29. This data suggested that these sia(−) isolates may harbor mutations in their covR/S genes. Based on the enhanced β-hemolysis which suggested possible CovR/S changes, we focussed attention on only these 15 sia(−) NT GBS isolates in our collection of sia(−) isolates for subsequent analysis.
Bacteria were streaked onto a 5% sheep blood agar plate and incubated for 24 h at 37 °C. The GBS COHI isolate was included as control which displayed a narrow zone of hemolysis
The microtitre hemolysis assay for sia(−) NT isolates
To confirm that the enhanced hemolytic activity and increase in orange pigmentation are reflected at the expression level in those isolates, we assayed cylE transcription. cylE (a gene encoded in cyl operon) is essential for GBS β-hemolytic production/orange pigmentation. RT-PCR using primers specific for the cylE gene revealed increases in cylE mRNA production in all sia(−) isolates assayed (average fold increase of 4.3 × ±2.3) (Supplemental Table 1). This suggested the enhanced β-hemolysis/orange pigment was due to increases in cyl operon transcription.
Assays for CAMP factor activity showed that these 15 isolates displayed no reaction or weaker CAMP activity on 5% sheep blood in comparison to the control isolate (COHI) (Supplemental Figure 2). Also, RT-PCR with primers specific for cfb, the gene responsible for the CAMP factor phenotypeCitation32, showed a reduction in cfb mRNA transcript in comparison to the control (Supplemental Table 1).
In addition to β-hemolytic activity/orange pigmentation production, and CAMP activity, we assayed the growth rates of the 15 sia(−) isolates in comparison to the GBS control isolate COHI in TH (an enriched medium) and RPMI (a minimal defined medium). In TH broth, the 15 sia(−) isolates displayed a faster growth rate compared to the control strain, with an average division time of 57 min (±0.01) compared to 64 min for COH1. In RPMI, all 15 sia(−) isolates with increased β-hemolytic activity were unable to grow in RPMI unlike the control which reached an average OD of 0.5 (±0.06) after 16 h of incubation.
To further demonstrate that the 15 sia(−) isolates did not produce polysaccharide capsule, we assayed for cpsE gene transcription. cpsE is a gene encoding a glycosyltransferase that initiates the biosynthesis of polysaccharide repeating units contained in the capsuleCitation33. Loss of cpsE gene expression can lead to lack of CPS expression. It was found that cpsE transcription was downregulated to very low levels in all sia(−) isolates in comparison to the COH1 control (Supplemental Table 1). This suggested the decrease in expression of the cpsE gene has led to loss of capsule expression in these GBS isolates.
Reduction in biofilm formation of the 15 sia(−) isolates
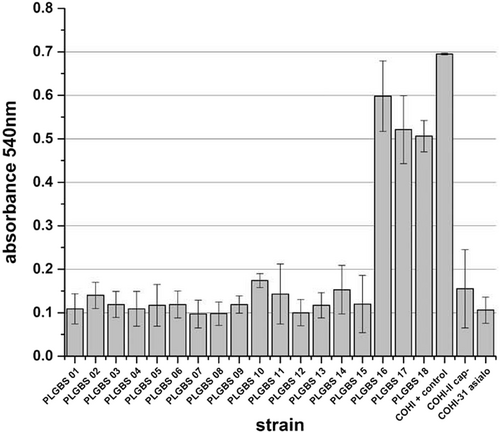
Previous research has shown that GBS CPS plays a major role in biofilm formation, which is important for GBS persistence and pathogenicityCitation15. We hypothesized that the sia(−) isolates in our collection would display a reduced ability to form biofilm. The unencapsulated and asialo mutants (negative controls) were impaired in their ability to form biofilm with an average absorbance at 600 nm of 0.2 (±0.09) and 0.1 (±0.03), respectively (Fig. ). The positive control, COHI, was able to form biofilm with an average absorbance at 600 nm of 0.695 (±0.002). GBS isolates that were sia(−) were unable to form biofilm having an average absorbance of 0.1 (±0.02, p > 0.001) (Fig. ).
Bacteria were grown at in RPMI supplemented with 1% glucose and 20% human plasma (HP). Cells were stained with crystal violet. Quantification was performed by solubilization of the stained biomass in ethanol/acetone (80/20) and measuring the absorbance at 540 nm. COHI was included as positive control while unencaspulated (COHI-II) and asialo (COHI-31-15) mutants were negative controls
Loss of resistance toward phagocytic killing by the 15 sia (−) isolates
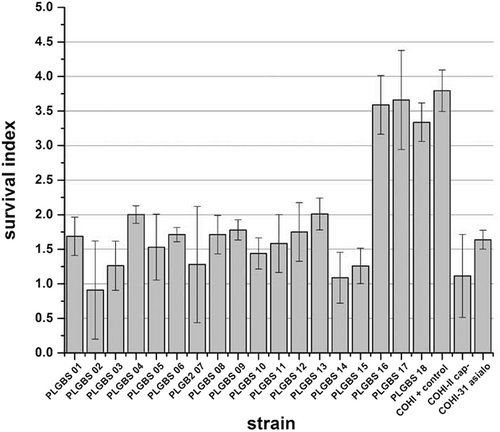
It has previously demonstrated that GBS CPS inhibits complement deposition, thereby reducing oposonophagocytotic clearance and contributing to immune resistanceCitation22. In addition, GBS isolates that display low hemolytic activity and high CPS expression have an increased resistance toward host immune defense. In contrast, isolates that exhibited high hemolytic activity and low capsule expression were phagocytosed in an oposonophagocytotic assayCitation25,Citation27. Based on these observations, we hypothesized that sia(−) isolates may be more susceptible to host immune defenses. To determine this, the survival of these GBS isolates in a human whole-blood killing assay was assessedCitation27,Citation30. The 15 sia(−) isolates displayed a low survival index with an average value of 1.5 (±0.4, p > 0.001) (ranged from 0.8 to 2) compared to the positive control (survival index value of 3.8 (±0.3)). The negative controls, the unencapsulated and asialo GBS mutants, had low survival indices of 1.1 (±0.6) and 1.6 (±0.1), respectively (Fig. ).
Bacteria (103 CFU/100 µl) were mixed with 300 µl of freshly drawn human blood in heparinised tubes, and incubated for 3 h with agitation at 37 °C, and dilutions were plated on blood agar for enumeration of CFU. Survival index was calculated as follows: (CFU at the end of the assay)/ (CFU at t = 0 h). GBS COHI was included as negative control while unencapsualted (COHI-II) and asialo (COHI-31-15) mutants were included as positive controls
Genetic analysis of the covR/covS genes of sia(−) NT isolates with increased β-hemolytic activity and orange pigment
Enhanced β-hemolytic activity, increased orange pigment production, loss of CAMP activity, inability to grow in minimal essential media, and loss of capsule expression strongly suggested mutations in the covR/S two-component regulatory system had occurred in these 15 sia(−) isolatesCitation25–Citation29. To verify whether the changes in the phenotypes of the 15 sia(−) isolates in this study was due to mutations that could potentially alter the amino acid coding sequence of CovR or CovS, the covR and covS genes were sequenced.
DNA sequencing of covR and covS genes identified mutations in the covR and covS sequences leading to the predicted amino acid changes shown in Table for 14 of the sia(−) isolates. PLGBS1 did not display any nucleotide changes in covR and covS genes. Eight of the 15 sia(−) isolates displayed mutations that predicated protein truncations of CovR (PLGBS2, 5, 8, 10, 11, 12, 13, and 15). Only three sia(−) isolates showed DNA sequence mutations in covS (PLGBS4, 12, and 14). Also, the DNA sequence of covS for PLGBS14 predicted an amino acid substitution of 21 amino acids in CovS. The remaining DNA sequence mutations predicted two–four amino acid substitutions in CovR (PLGBS3, 6, 7, and 9) (Table ). As PLGBS1 did not display any changes in the nucleotide sequences of covR and covS, it is likely other genes are involved in lack of CPS expression.
The predicted amino acid changes in CovR and CovS based on nucleotide mutation(s) in the covR and covS genes identified among 15 sia(−) NT GBS isolates
Sia(+) isolates
Knowing that the presence of surface sialic acid can predict the presence of polysaccharide capsule, it was unusual to find in our collection three GBS isolates (PLGBS16, PLGBS17, and PLGBS18) which failed to react with antisera raised against the nine known CPS types (Ia, Ib, and II–VIII) yet were sialic acid positiveCitation23 (data not shown). Additionally, these three isolates could not be genotyped by our previously described RT-PCR typing assay (which includes Ia, Ib, and II–IX)Citation23, suggesting novel mechanisms of encapsulation. A polysaccharide stain revealed that these three GBS isolates microscopically displayed CPS surrounding the cell wall (Supplemental Figure 3). These observations prompted us to further analyze these three GBS isolates with unidentifiable polysaccharide capsules.
Phenotypic properties of GBS sia(+) NT GBS isolates
PLGBS16, PLGBS17, and PLGBS18 isolates exhibited β-hemolytic activity similar to the wild-type control, COHI (Fig. ), grew as a white colony (Supplemental Figure 1), and displayed enhanced CAMP activity (Supplemental Figure 2). Growth rate comparisons in TH (enriched media) and RPMI (minimal media) of the three sia(+) NT GBS isolates compared to the control GBS COHI isolate showed that they have similar growth rates with division times of 66 (PLGBS16), 65 (PLGBS17), and 63 (PLGBS18) minutes compared to 64 min for COHI. The growth profile for the three isolates in RPMI were similar to the control with an average OD of 0.55 (±0.12), 0.59 (±0.09), and 0.6 (±0.06), respectively, after 16 h of incubation.
Biofilm formation by sia(+) NT GBS isolates
Based on the important role of GBS capsule in biofilm formationCitation15, we hypothesized that the three sia(+) GBS isolates were able to form biofilm unlike the 15 previously characterized sia(−) isolates. All three isolates were able to form a biofilm in RPMI media containing 20% human plasma and 1% glucoseCitation15,Citation34–Citation36 (average absorbance at 600 nm of 0.5 (±0.05)) similar to the positive control COHI (0.7, ±0.002) (Fig. ). Negative controls (the unencapsulated and asialo mutants) were impaired in their ability to form biofilms with an average absorbance at 600 nm of 0.2 (±0.09) and 0.1 (±0.03), respectively (Fig. ).
Resistance of phagocytic killing by sia(+) NT GBS isolates
To assay resistance to phagocytic killing, the ability of the three sia(+) NT GBS isolates to survive in fresh human blood was assayed in a human whole-blood killing assayCitation27,Citation30. The three isolates displayed high-survival indices with an average index of 3.5 (±0.2) similar to the positive control COHI strain (survival index 3.8, ±0.3) (Fig. ), indicating that three sia(+) isolates were able to resist killing likely due to the presence of the capsule.
Genetic analysis of sia(+) NT GBS isolates
As PLGBS16, PLGBS17, and PLGBS18 represented potentially new GBS CPS variant(s), whole-genome sequence analyses were done focusing on the MLST and cps gene comparative analysis. MLST data for PLGBS16, PLGBS17, and PLGBS18 were assigned a ST1 designation for PLGBS16 and PLGBS17 that were included in clonal complex (CC) 1. ST1/CC1 (http://pubmlst.org/sagalactiae/) has been reported to be commonly found among CPSV isolatesCitation37. PLGBS18 was assigned a ST2/CC1 designation.
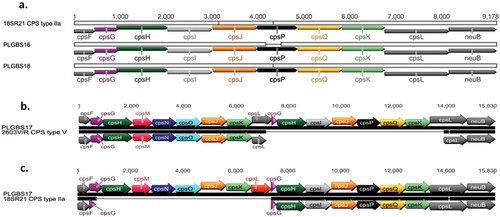
To investigate whether isolates PLGBS16, PLGBS17, and PLGBS18 encode novel genes involved in CPS synthesis, we compared the DNA sequences of the cps gene clusters of these isolates to the cps gene cluster of known CPS typesCitation33. Comparative analysis of the cps gene cluster revealed that PLGBS16 and PLGBS18 and the previously sequenced cps2a gene cluster (18SR21, GenBank accession #AAJO01000077) shared highly homologous sequences across the cps locus (Fig. and Table ). Poyart et al. have previously suggested that CPSII is encoded by two cps clusters, with suggested designation subtypes IIa and IIb represented by 18SR21 and AY375362 cps sequencesCitation38. PLGBS16 and PLGBS18 shared low homology with cps sequences of CPSIIb (GenBank accession #AY375362) (Table ). A CPS dot blot assay using earlier prepared in-house rabbit antibody raised against CPSII did not identify these isolates as CPSII, suggesting that the in-house antibody preparation targeted CPSIIb only. In contrast to PLGBS16 and PLGBS18, the isolate-designated PLGBS17 isolate shared highly homologous sequences with CPSIIa (18SR21) and CPSV (2603V/R) (Fig. and Table )Citation38,Citation39. Two well-conserved regions spanning from cpsA to cpsF and from neuB to neuA, flanking a central region between cpsG and cpsL, were found in PLGBS17 (Fig. ). In the central region, the PLGBS17 isolate showed a high similarity to CPS-determining region of CPS type V, including the following genes cpsG, cpsH, cpsM, cpsN, cpsO, and cpsL. It also shared high similarity to cps2a-specific genes including a fragment of cpsG (228 bp), and cpsH, cpsI, cpsQ, cpsS, and a fragment of cpsL (725 bp) (Fig. and Table ). These results suggest that cps-specific region of PLGBS17 is a hybrid of CPSIIa and V. Further analysis using in-house-prepared rabbit polyclonal antibodies raised against CPSII and commercial antibodies against CPSV failed to react in a CPS dot blot assay (data not shown). Together, the data suggested a novel hybrid CPS type composed of cps2a and cps5 genes.
a 18SR21 (CPSIIa) vs. PLGBS16 and PLGBS18, b PLGBS17 vs. 2603V/R (CPSV), and c PLGBS17 vs. 18SR21 (CPSIIa)
Nucleotide sequence identity comparisons for individual cps genes for the 3 sia(+) PLGBS16, PLGBS17, and PLGBS18 vs the control type strains (18SR21 CPSIIa, AY375362 CPSIIb and AE009948 CPSV)
Discussion
The polysaccharide capsule is an important virulence factor of GBS and a convenient target for typing to aid in the understanding of GBS epidemiology. GBS strains for which a polysaccharide type cannot be determined by serological assays are usually reported as a nontypable as they typically do not express a capsule. The use of molecular assays targeting cps genes in GBS have aided in identifying the cps genes in these nontypable GBS isolates thereby assigning a GBS type even though no capsule may be expressed. In addition to lack of capsule expression, a subset of GBS that are nontypable may actually express a capsule that is unrecognizable using serological or molecular assays. Identifying and understanding the prevalence of those GBS strains that do express CPS yet are considered NT can provide information regarding disease trends of these unencapsulated yet invasive bacteria.
To better understand the NT phenotype of GBS isolates, we undertook a detailed characterization of a collection of nontypable GBS collected from cases of invasive disease in Alberta. For the sia(−) isolates, we focussed on potential changes in GBS that were likely caused by changes in the two-component GBS global regulatory system CovR/S. As CovR/S controls the expression (or lack of) of an array of phenotypes, we focussed only on those isolates that were hyperhemolytic/hyperpigmented and were CAMP factor negative. These phenotypic attributes plus loss of biofilm formation and susceptibility to phagocytic killing strongly suggested changes had occurred in the CovR/S system. DNA sequencing of both the covR and covS genes identified an array of mutations that had occurred in these genes.
A number of covR/S mutations were discovered in 14 out of the 15 hyperhemolytic NT sia(−) isolates. It is interesting that half of the covR/S mutations (7/14), potentially resulted in truncations of CovR at amino acid number 2. This suggests that there was a loss of almost the entire CovR protein in these GBS isolates and serves as a logical explanation for loss of capsule production. As this mutation occurred frequently in our collection, it is possible this represents a non-encapsulated clone circulating in the Alberta population. However, a close examination of the years in which these isolates were collected from cases of iGBS disease shows a wide-temporal period reducing the likelihood of the isolates being clonal.
With respect to CovS, three isolates were found to have DNA sequence mutations in the covS coding sequence potentially resulting in CovS amino acid changes. One change in particular was the substitution of a 21 amino-acid-long stretch of amino acids located at the C-terminal end of CovS (PLGBS14). covR or covS mutations are not unusual as a variety of mutations in these genes have been previously reported. Whidbey et al. previously reported a hyperhemolytic/hyperpigmented GBS with a Glu120Pro substitution in CovRCitation40. Also, work by Almeida et al. reported a collection of mutations in the covS gene. These were primarily single amino acid substitutions (Thr43Ile, Ala87Val, Gly133Ser, His209Asn, Trp297Leu, and Glu337Ser) and the one double substitution (Trp297Cys and Gly298Trp)Citation41. None of these amino acid changes were predicted amino acid changes in the covR and covS mutations found in our isolates suggesting mutations of covR and covS may occur in multiple locations in these genes. Understanding the frequency of covR/S mutants as they occur is important as this two-component regulatory system coordinates a number of virulence factors including vaccine targets. It is unclear if covR/S mutants would be favored if a GBS vaccine is introduced that contains one or more virulence factors that can be downregulated by mutations in covR/S, such as CPS.
The finding of three GBS sia(+) isolates with non-serological and non-PCR typable cps gene clusters was unexpected as it has been established that there are only ten known CPS variants designated Ia, Ib, and II–IX. However, based on this study and the work of others, it is clear that a second cps2 gene cluster exists for GBS isolates from cases of invasive disease in humansCitation38. The small number of reports for CPSIIa in the literature suggests that CPSIIa is relatively rare in occurrence possibly explaining why it has not been classed as one of the well-established CPS types. Differences between the two cps2 gene clusters is evident in the literature; however, a clear recognition that two CPSII capsules exist is generally lacking. Berti et al. have previously compiled a nucleotide sequence identity table for all ten CPS types listing each cps geneCitation19. For CPSII, these were reported as cpsA, cpsB, cpsC, cpsD, cpsE, cpsF, cpsG, cpsH, cpsI, cpsJ, and cpsK. Recently, Kapatai et al. compared the cps loci of all ten serotypesCitation42. In this report, the CPSII genes were designated cpsA, cpsB, cpsC, cpsD, cpsE, cpsF, cpsG, cpsH, cpsI, cpsJ, cpsP, cpsQ, and cpsK. The major difference between the two gene clusters being the presence of cpsP and cpsQ in one report and absent in the other. It is highly likely CPSIIb (no cpsP and no cpsQ) is the more common CPSII type and is probably the CPSII serotyped by most laboratories performing GBS serotyping. To be accurate, GBS CPS typing reports should split CPSII types into IIa and IIb even though CPSIIa is likely uncommon. This would extend the GBS CPS typing collection to 11 types (Ia, Ib, IIa, IIb, III, IV, V, VI, VII, VIII, and IX). The genes cpsP and cpsQ from the cps2a gene cluster could be easily incorporated into PCR-based CPS typing schemes allowing investigators to readily identify CPSIIa types.
It was interesting that even though CPSIIa is uncommon, a cps2a/cps5 hybrid gene cluster was discovered in our nontypable collection. This cps2a/cps5 hybrid (genes cpsH, cpsI, cpsQ, cpsS, cpsK, and a fragment of cpsL from cps2a+ genes cpsG, cpsH, cpsN, cpsM, cpsO, cpsK from cps5) failed to react with either CPSII antisera (now known to be CPSIIb antisera) or CPSV antisera from our antisera collection. The finding of only one isolate with this hybrid cps gene sequence suggests that this is a rare event and likely does not give much selective advantage over other cps types, otherwise more isolates with this CPS type would have likely been found.
In summary, characterization of a collection of NT sia(−) isolates found these bacteria did not express a variety of phenotypic traits previously shown to be regulated by covR/S. Also, characterization of the NT sia(+) isolates found two isolates that contained a cps gene cluster designated cps2a and an isolate that contained a novel hybrid cps gene cluster comprised of genes from the cps2a and cps5 gene clusters.
Materials and methods
Bacterial strains, growth conditions, and oligonucleotides
The relevant characteristics of the bacterial strains used in this study are listed in Table . We reported previously that 159/1683 GBS isolates collected from patients between 2003 and 2013 were identified as NTCitation31 and of those, 84 of the 159 were sialic acid negative (sia(−))Citation23, whereas 75/159 were sialic acid positive (sia(+))Citation23. Of the 75 serologically nontypable sia(+) isolates, only three isolates (PLGBS16, PLGBS17, and PLGBS18) could not be genotyped by our previously described RT-PCR typing assay (which included Ia, Ib, and II–IX)Citation23. All GBS isolates were cultured in TH broth or Columbia blood agar plates (Dalynn Biologicals, Calgary, Canada) containing 5% sheep blood or RPMI 1640 (Thermo Fisher Scientific, Toronto, Canada) as a synthetic medium. GBS liquid cultures were grown in standing-filled flasks. All incubations were at 37 °C.
Strains used in the study
Assay for hemolytic activity
GBS hemolytic activity was assayed as previously describedCitation27. Briefly, an overnight culture (109 cfu) in TH broth were centrifuged for 5 min at 3000 × g, washed twice with phosphate-buffered saline (PBS), and re-suspended in 1 ml of PBS. In a 96-well conical-bottom microtiter plate (MP Biomedicals, Santa Ana, USA), 100 µl per well (108 cfu) of the bacterial resuspension was placed in the first well, and serial twofold dilutions in PBS were performed across the plate, each in a final volume of 100 µl. An equal volume of 1% sheep erythrocytes washed once with PBS (5 min by centrifugation at 3000 × g to avoid non-specific red blood cell lysis), and re-suspended in PBS, was then added to each well, and the plate was incubated at 37 °C for 60 min.
PBS alone and 0.1% SDS were used as negative and positive controls for hemolysis, respectively. After incubation, the plates were centrifuged at 3000 × g for 10 min to pellet the unlysed red blood cells and GBS, and 100 µl of the supernatant was transferred to a replica plate. Hemoglobin release was assessed by measuring A420, and the hemolytic capacity of a given strain was determined. Briefly, the positive control absorbance value (0.1% SDS) and absorbance values of each isolate were subtracted from the negative control value (PBS) (normalized values). The normalized values of each isolate were divided by the normalized positive control absorbance value and multiplied by 100. This value was designated as the hemolytic capacity.
All assays were performed in triplicate, repeated three times, and the mean value ± standard deviation (SD) is indicated.
Assay for orange pigment production
The broth cultures were incubated to the stationary phase of growth (18 h) and then centrifuged. The pellet was spotted onto nitrocellulose membrane (20 µl/spot) using a bio-dot apparatus (Bio-Rad, Hercules, USA), which was dried for 30 min at room temperature.
RNA isolation
THB broth (20 ml) was inoculated (1:20) with an overnight culture of GBS strains and incubated at 37 °C. Exponentially growing cells (OD600 0.3–0.4) were harvested for 2 min at 6000 × g at 4 °C. The pellets were re-suspended by vortexing in 400 ml of resuspension buffer (12.5 mM Tris, 5 mM EDTA and 10% glucose). The supernatant was transferred to a fresh tube, and 1 ml of Trizol reagent (Thermo Fisher Scientific, Toronto, Canada) was added. The sample was incubated for 5 min at room temperature. Total RNA was extracted twice with chloroform-isoamyl alcohol (24:1, v/v) and precipitated in 0.7 volumes of isopropanol. After a washing step with 70% ethanol, the RNA pellet was dissolved in sterile DNase and RNase-free water (Thermo Fisher Scientific, Toronto, Canada) and treated for 30 min at 37 °C with RNase-free DNase I (1 unit per mg of total RNA) in 50 mM Tris-HCl (pH 7.5) and 10 mM MnCl2. DNase was inactivated by phenol–chloroform extraction, and the RNA was precipitated and washed with 70% ethanol, re-dissolved in RNase-free water and quantified by absorbance at 260 and 280 nm. Purity and integrity of RNA were controlled on agarose gels, and RNA was stored at −20 °C until use.
Detection of mRNA by RT-PCR
Using a high capacity cDNA reverse transcription kit with RNAse inhibitor (Thermo Fisher Scientific, Toronto, Canada), 0.8 µg RNA was reverse transcribed as recommend by manufacture. RT-PCRs were carried out by using Fast SYBR® Green Master Mix (Thermo Fisher Scientific, Toronto, Canada). Reactions were carried out in a final volume of 20 ml containing 0.5 µg of RNAs, 0.5 μM of each forward and reverse primer (Integrated DNA Technology, IDT, USA) for each cpsE, cylE, cfb, and rpsL and 10 µl of master mix. The real-time PCR was performed using a TaqMan RT-PCR Applied Biosystems System 7500 (Applied Biosystems, Culver City, USA). The cycling conditions were denaturation at 95 °C for 1 min, followed by 40 cycles of amplification at 95 °C for 3 s, 60 °C for 30 s. The rate of temperature increase was 1 °C/s (or 0.5 °C/s), and fluorescence was acquired once. Each RT-PCR was repeated three times. The list of oligonucleotides used in this study is indicated in Supplementary Table 1. The differences between the genes cylE, cpsE, and cfb (genes assayed) vs. the housekeeping gene (rpsL) in NT sia(−) isolates and the same genes in the control COHI strain were calculated. These values were subtracted from each other (test – control, (ΔCTE − ΔCTC)) to determine the ddCT value. Fold change was calculated based on log2-ddCT. The average and standard deviation of the fold change were calculated from three independent experiments.
Genomic DNA extraction
Genomic DNA extraction was performed as follows. Overnight broth cultures (1.5 ml) were centrifuged for 10 min at 3000 × g. Genomic DNA was re-suspended in 500 µl of 1× PBS (8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4, and 1000 ml H2O [pH 7.2]) and washed two times with PBS. The pellet was used to extract genomic DNA using the Qiagen DNA mericon kit (Qiagen, Dusseldorf, Germany). Extracted genomic DNA was concentrated and dissolved in 30 µl Qiagen elution buffer or water and stored at −20 °C. RNase pretreatment was done prior to quantification of genomic DNA.
PCR amplification and sequencing of covR and covS
PCR assays that target either covR or covS based on the sequences from GBS strain 2603V/R (Genbank accession #AE009948) were developed (Supplemental Table 2). The amplified fragments were sequenced using the same primers, covR-F and covR-R or covSA-F and covSA-R and covSB-F and covSB-R (Integrated DNA Technology, Coralville, USA) (Supplementary Table 1) used for amplification and compared to the GBS genome (Genbank accession #AE009948) available in Genbank.
Biofilm forming assay
The biofilm forming assay was performed as described previouslyCitation15. Briefly, GBS were grown in TH (Becton Dickinson, Franklin Lakes, USA) broth. Human plasma was collected from a human volunteer. Overnight GBS cultures were used to inoculate RPMI-glucose or RPMI-glucose supplemented with human plasma at an OD600 of 0.1, vortexed briefly and 180 µl volumes dispensed in 96 wells plate (MP Biomedicals, USA) and incubated for 18 h at 37 °C. The OD600 of each culture was measured to ensure that all cells had reached stationary phase with a similar density, and the cells were washed twice in PBS and air-dried for 15 min. Biofilms were stained with 0.4% crystal violet for 30 min and the wells were washed twice with PBS and air-dried. The bacterial biomass was then re-suspended for quantification in ethanol/acetone (80/20) solution and OD540 was measured. When OD values were above 1, twofold dilutions were performed for accuracy. The assay was performed in triplicate and at least three independent experiments were performed.
Human whole-blood killing assay
Human whole-blood killing assay was performed as described previouslyCitation27,Citation30. Briefly, GBS was grown to early logarithmic phase, washed, and re-suspended in PBS. Inocula of 103 CFU in 100 μl were mixed with 300 μl of freshly drawn human blood in heparinised tubes, and incubated for 3 h with agitation at 37 °C, and dilutions were plated on blood agar for enumeration of CFU. The survival index was calculated as follows: (CFU at the end of the assay)/(CFU at t = 0 h). The assay was performed in triplicate and repeated two times independently.
Whole-genome sequencing
PLGBS16, PLGBS17, and PLGBS18 isolates were sequenced using a MiSeq instrument with an average sequencing depth of 120× (Illumina, San Diego, USA). Library preparation was performed using a tagmentation process. One nanogram genomic DNA as input and the Illumina Nextera XT Library preparation kit (FC-131-1096) as per manufacturer’s protocol (Illumina, San Diego, USA) was used. Resultant libraries were purified from free primers using Ampure beads (1:0.8 reaction mix:bead ratio). Libraries were qualified using the Agilent Bioanalyzer (Agilent, Santa Clara, USA) and quantified using a Qubit fluorimeter and Qubit HS DNA reagents (Thermofisher Scientific, Toronto, Canada). Libraries were loaded at 10 pM on an Illumina MiSeq v2 reagent kit (MS-102-2003) and processed at 2 × 500 cycles with 1% PhiX control DNA (FC-110-3001). Sequence data for the three genomes have been deposited in the NCBI genome database (BioProject PRJNA433769). Illumina reads were assembled to contigs by CLC genomic bench work using de novo assembly. The cps operons of PLGBS16 and PLGBS18 were assembled in one contig, whereas the cps locus of PLGBS17 was built from five different contigs encoding cps genes. The assembled genome was annotated using online annotation websites Rapid Annotation of Prokaryotes using Subsystem (RAST)Citation43. Contigs corresponding to the chromosomal region encompassing the cps operon were identified and aligned by Muscles using Geneious and by using as query sequence the sequence of strain 18SR21 (GenBank accession ##AAJO01000077)Citation32 or 2603V/R (GenBank accession# AE009948)Citation33 cps region.
Multilocus sequence typing (MLST) assay and assignment to clonal clusters
Multilocus sequence typing (MLST) was carried out as described previouslyCitation44. Briefly, the seven housekeeping genes (adhP, pheS, atr, glnA, sdhA, glcK, and tkt) were assigned an allele number based on their sequences for the isolates sequenced. Each isolate was assigned a sequence type (ST) based on the allelic profile of the seven amplicons for each strain were grouped into CCs using the eBURST software programCitation45,Citation46. eBURST was set at the default setting that identified groups of related STs using the most stringent (conservative) definition. All members assigned to the same group shared identical alleles at six of the seven loci with at least one other member of the group (http://pubmlst.org/sagalactiae/).
Double immunodiffusion assay for GBS CPS typing
CPS typing was performed using the Lancefield heat-acid extraction followed by a double immunodiffusion method as described previouslyCitation47,Citation48 for sia(+) isolates, PLGBS16, PLGBS17, and PLGBS18. The immunodiffusion assay of GBS CPS typing used for this study was based on reactions with antisera raised against cps types Ia, Ib, II, III, IV, V, VI, VII, and VIII. The type-specific antisera panel was prepared in rabbits within the laboratoryCitation47,Citation48.
CPS dot blot assay
A dot blot assay to detect CPS was performed as previously described with minor modificationsCitation49 for the sial(+) NT GBS isolates, PLGBS16, PLGBS17, and PLGB18. Late exponentially growing bacteria were washed in phosphate-buffered saline (PBS) and re-suspended in PBS to give an optical density at 600 nm of ~2 (Beckman Coulter, Mississauga, Canada). The bacterial suspension was spotted onto nitrocellulose membrane (20 µl/spot) using a bio-dot apparatus (Bio-Rad, USA), which was dried for 30 min at room temperature. The membranes were washed for 15 min with TBS (6.05 g Tris, 8.76 g NaCl in 1000 ml of H2O [pH 7.5]), incubated with blocking buffer (5% skim milk, 0.1% Tween 20 in TBS) for 60 min at 37 °C. The membrane was subsequently washed with TBS for 15 min. CPS was detected using specific rabbit polyclonal antibodies raised against CPSII (in-house preparation), CPSIII, or CPSV (Statens Serum Institute, Copenhagen, Denmark) at 1:1000 dilution. The secondary horseradish peroxidase-coupled anti-rabbit secondary antibody (Promega, Madison, USA) was used at 1:50,000 dilution. The membrane was washed 3 × 15 min with TBST and 2 × 15 min with TBS before developing with SigmaFast BCIP/NBT (Sigma-Aldrich, St. Louis, USA) for ~5 min. Development was stopped using three changes of distilled water.
Supplementary Information
Download MS Word (16.3 KB)Supplementary Figure S1
Download JPEG Image (186.1 KB){kind=link}
Supplementary Information
Download MS Word (199.5 KB)Supplementary Figure S2
Download JPEG Image (185.5 KB){kind=link}
Acknowledgements
We gratefully acknowledge the diagnostic laboratories in Alberta who identified and submitted for CPS typing GBS isolates from cases of invasive disease.
Authors' contributions
A.A. conceptualized and performed the experiments as well as participated in writing the manuscript. G.J.T. conceptualized and participated in writing the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41426-018-0138-6).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Research Data
References
- SchuchatAGroup B StreptococcusLancet1999353 51 5610.1016/S0140-6736(98)07128-1
- BakerCJBarrettFFTransmission of group B streptococci among parturient women and their neonatesJ. Pediatr.19738391992510.1016/S0022-3476(73)80524-4
- BakerCJBarrettFFGroup B streptococcal infections in infants. The importance of the various serotypesJAMA19742301158116010.1001/jama.1974.03240080040025
- BerardiAGroup B streptococcus late-onset disease: 2003-2010Pediatrics2013131e361e36810.1542/peds.2012-1231
- CampbellJRGroup B streptococcal colonization and serotype-specific immunity in pregnant women at deliveryObstet. Gynecol.200096498503
- EdmondKMGroup B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysisLancet201237954755610.1016/S0140-6736(11)61651-6
- FranciosiRAKnostmanJDZimmermanRAGroup B streptococcal neonatal and infant infectionsJ. Pediatr.19738270771810.1016/S0022-3476(73)80604-3
- TapsallJWPhillipsEAPresumptive identification of Group B streptococci by rapid detection of CAMP factor and pigment productionDiagn. Microbiol. Infect. Dis.1987722522810.1016/0732-8893(87)90010-1
- RamaswamySVIdentification of novel cps locus polymorphisms in nontypable Group B streptococcusJ. Med. Microbiol.20065577578310.1099/jmm.0.46253-0
- DoranKSNizetVMolecular pathogenesis of neonatal Group B streptococcal infection: no longer in its infancyMol. Microbiol.200454233110.1111/j.1365-2958.2004.04266.x
- HerbertMABeveridgeCJSaundersNJBacterial virulence factors in neonatal sepsis: Group B streptococcusCurr. Opin. Infect. Dis.20041722522910.1097/00001432-200406000-00009
- RubensCEWesselsMRKuypersJMKasperDLWeiserJNMolecular analysis of the two Group B streptococcal virulence factorsSemin. Perinatol.1990142229
- SlotvedHCKongFLambertsenLSauerSGilbertGLSerotype IX, a proposed new streptococcus agalactiae serotypeJ. Clin. Microbiol.2007452929293610.1128/JCM.00117-072045254
- AfsharBInternational quality assurance for laboratory identification and typing of Streptococcus agalactiae (Group B streptococci)J. Clin. Microbiol.2011491475148210.1128/JCM.02365-103122801
- XiaFDCapsular polysaccharide of Group B streptococcus mediates biofilm formation in the presence of human plasmaMicrobes Infect.201517717610.1016/j.micinf.2014.10.007
- HarrisonLHSerotype distribution of invasive Group B streptococcal isolates in Maryland: implications for vaccine formulationJ. Infect. Dis.1998177998100210.1086/515260
- ChaffinDOMenteleLMRubensCESialylation of Group B streptococcal capsular polysaccharide is mediated by cpsK and is required for optimal capsule polymerization and expressionJ. Bacteriol.20051874615462510.1128/JB.187.13.4615-4626.20051151781
- WesselsMRHaftRFHeggenLMRubensCEIdentification of a genetic locus essential for capsule sialylation in type III group B streptococciInfect. Immun.199260392400257641
- BertiFStructure of the type IX group B streptococcus capsular polysaccharide and its evolutionary relationship with types V and VIIJ. Biol. Chem.2014289234372344810.1074/jbc.M114.5679744156066
- CieslewiczMJStructural and genetic diversity of Group B streptococcus capsular polysaccharidesInfect. Immun.2005733096310310.1128/IAI.73.5.3096-3103.20051087335
- CarlinAFGroup B streptococcus suppression of phagocyte functions by protein-mediated engagement of human Siglec-5J. Exp. Med.20092061691169910.1084/jem.200906912722167
- MarquesMBKasperDLPangburnMKWesselsMRPrevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III Group B streptococciInfect. Immun.19926039863993257427
- AlhhazmiAPandeyATyrrellGJidentification of the Group B streptococcus capsule type using a dual phenotypic/genotypic assayJ. Clin. Microbiol.2017552637265010.1128/JCM.00300-175648701
- Di PaloBAdaptive response of Group B streptococcus to high glucose conditions: new insights on the CovRS regulation networkPLoS ONE20138e6129410.1371/journal.pone.00612943621830
- LamyMCCovS/CovR of Group B streptococcus: a two-component global regulatory system involved in virulenceMol. Microbiol.2004541250126810.1111/j.1365-2958.2004.04365.x
- LupoARuppenCHemphillASpellerbergBSendiPPhenotypic and molecular characterization of hyperpigmented Group B StreptococciInt. J. Med. Microbiol.201430471772410.1016/j.ijmm.2014.05.003
- SendiPBacterial phenotype variants in Group B Streptococcal toxic shock syndromeEmerg. Infect. Dis.20091522323210.3201/eid1502.0809902657631
- Whidbey, C. et al. A hyperhaemolytic/hyperpigmented Group B streptococcus strain with a CovR mutations isolated from an adolescent patient with sore throat. Clin. Res. Infect. Dis.2, 1018 (2015).
- WhidbeyCA hemolytic pigment of Group B streptococcus allows bacterial penetration of human placentaJ. Exp. Med.20132101265128110.1084/jem.201227533674703
- LiuGYSword and shield: linked Group B streptococcal beta-hemolysin/cytolysin and carentoid pigment function to subvert host phagocyte defenseProc. Natl Acad. Sci. USA2004101144911449610.1073/pnas.0406143101
- AlhhazmiAHurteauDTyrrellGJEpidemiology of invasive Group B streptococcal disease in Alberta, Canada 2003-2013J. Clin. Microbiol.2016541774178110.1128/JCM.00355-164922093
- KeDDevelopment of conventional and real-time PCR assays for the rapid detection of Group B streptococciClin. Chem.200046324331
- CieslewiczMJKasperDLWangYWesselsMRFunctional analysis in the type Ia Group B streptococcus cluster of genes involved in extracellular polysaccharide production by diverse species of streptococciJ. Biol. Chem.200127613914610.1074/jbc.M005702200
- D’UrzoNAcidic pH strongly enhances in vitro biofilm formation by a subset of hypervirulent ST-17 Streptococcus agalactiae strainsAppl. Environ. Microbiol.2014802176218510.1128/AEM.03627-133993151
- ParkerREAssociation between genotypic diversity and biofilm production in Group B streptococcusBMC Microbiol.2016168610.1186/s12866-016-0704-94875601
- RosiniRMargaritIBiofilm formation by Streptococcus agalactiae: influence of environmental conditions and implicated virulence factorsFront. Cell. Infect. Microbiol.201546
- NeemuchwalaATeateroSAtheyTBMcGeerAFittipaldiNCapsular switching and other large-scale recombination events in invasive sequence Type 1 group B streptococcusEmerg. Infect. Dis.2016221941194410.3201//eid2211.1520645088006
- PoyartCMultiplex PCR assay for rapid and accurate capsular typing of Group B streptococciJ. Clin. Microbiol.2007451985198810.1128/JCM.00159-071933079
- TettlinHComplete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiaeProc. Natl Acad. Sci. USA200299123911239610.1073/pnas.182380799
- WhidbeyCA hyperhemolytic/hyperpigmented Group B streptococcus strain with a CovR mutation isolated from an adolescent patient with a sore throatClin. Res. Infect. Dis.2015210184762654
- AlmeidaAParallel evolution of group B streptococcus hypervirulent clonal complex 17 unveils new pathoadaptive mutationsmSystems20172e000741710.1128/mSystems.00074-175585690
- KapataiGPatelDEfstratiouAChalkerVJComparison of molecular serotyping approaches of Streptococcus agalactiae from genomic sequencesBMC Genomics20171810.1186/s12864-017-3820-55455115
- OverbeekRThe SEED and the rapid annotation of microbial genomes using subsystems technology (RAST)Nucleic Acids Res.201442D206D21410.1093/nar/gkt1226
- JonesNMultilocus sequence typing system for Group B streptococcusJ. Clin. Microbiol.2003412530253610.1128/JCM.41.6.2530-2536.2003156480
- FeilEJLiBCAanensenDMHanageWPSprattBGeBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing dataJ. Bacteriol.20041861518153010.1128/JB.186.5.1518-1530.2004344416
- SprattBJHanageWPAanensenDMFeilEJDisplaying the relatedness among isolates of bacterial species – the eBURST approachFEMS Microbiol. Lett.200424112913410.1016/j.femsle.2004.11.015
- JohnsonDRFerrieriPGroup B streptococcal Ibc protein antigen: distribution of two determinants in wild-type strains of common serotypesJ. Clin. Microbiol.198419506510271105
- LancefieldRCMcCartyMEverlyWNMultiple mouse-protective antibodies directed against Group B streptococci. Special reference to antibodies effective against protein antigensJ. Exp. Med.197514216517910.1084/jem.142.1.165
- BellaisSCapsular switching in Group B streptococcus CC17 hypervirulent clone: a future challenge for polysaccharide vaccine developmentJ. Infect. Dis.20122061745175210.1093/infdis/jis605
- RubensCEHeggenLMHaftRFWesselsMRIdentification of cpsD, a gene essential for type III capsule expression in Group B streptococciMol. Microbiol.1993884385510.1111/j.1365-2958.1993.tb01631.x
- RubensCEWesselsMRHeggenLMKasperDLTransposon mutagenesis of type III Group B streptococcus: correlation of capsule expression with virulenceProc. Natl Acad. Sci. USA1987847208721210.1073/pnas.84.20.7208
- TyrrellGJKennedyAShokoplesSESherburneRKBinding and invasion of HeLa and MRC-5 cells by Streptococcus agalactiaeMicrobiology20021483921393110.1099/00221287-148-12-3921
- JiangSMCieslewiczMJKasperDLWesselsMRRegulation of virulence by a two-component system in Group B streptococcusJ. Bacteriol.20051871105111310.1128/JB.187.3.1105-1113.2005545708