
These authors contributed equally: Sascha Knauf, Jan F. Gogarten, Verena J. Schuenemann and Hélène M. De Nys.
Deceased: Emmanuel K. Batamuzi.
These authors also contributed equally: David Šmajs, Kay Nieselt, Johannes Krause and Sébastien Calvignac-Spencer.
Dear Editor,
The bacterium Treponema pallidum (TP) causes human syphilis (subsp. pallidum; TPA), bejel (subsp. endemicum; TEN), and yaws (subsp. pertenue; TPE)Citation1. Although syphilis has reached a worldwide distributionCitation2, bejel and yaws have remained endemic diseases. Bejel affects individuals in dry areas of Sahelian Africa and Saudi Arabia, whereas yaws affects those living in the humid tropicsCitation1. Yaws is currently reported as endemic in 14 countries, and an additional 84 countries have a known history of yaws but lack recent epidemiological dataCitation3,Citation4. Although this disease was subject to global eradication efforts in the mid-20th century, it later reemerged in West Africa, Southern Asia, and the Pacific regionCitation5. New large-scale treatment options triggered the ongoing second eradication campaign, the goal of which is to eradicate yaws globally by 2020Citation5.
TPE is typically considered to be a strictly human pathogen, a perception that may partially have arisen from a lack of detailed data on nonhuman primate (NHP)-infecting treponemes. Indeed, a number of African NHPs show skin ulcerations that are suggestive of treponemal infections, and antibodies against TP have been detected in wild NHP populationsCitation6,Citation7. Although genetic studies confirmed that monkeys and great apes are infected with TP strainsCitation8–Citation10, most of these analyses only used short DNA sequences. Thus, the small number of examined polymorphic sites largely precluded assignment of these strains to a particular TP subspeciesCitation9, especially considering that sporadic recombination events between subspecies have been reportedCitation11. The only simian strain whose whole genome has been sequenced (Fribourg-Blanc, isolated from a Guinea baboon (Papio papio) in 1966Citation7) unambiguously clustered with human-infecting TPE strainsCitation12.
A fundamental question with regard to yaws evolution, and possibly yaws eradication, is whether humans and NHPs are commonly infected with the same pathogen (TPE) and whether transmission between NHPs and humans occurs. To determine which pathogen causes treponematoses in NHPs across sub-Saharan Africa, we collected samples from symptomatic wild individuals belonging to three NHP species (Cercocebus atys, Chlorocebus sabaeus, and Papio anubis) from four independent populations in West and East Africa (, Supplementary Table S1, Supplementary Materials). Samples were collected from NHPs at Taï National Park (TaïNP; Côte d’Ivoire), Bijilo Forest Park (BFP, the Gambia), Niokolo-Koba National Park (NKNP, Senegal), and Lake Manyara National Park (LMNP, Tanzania). Monkeys presented yaws-like orofacial and limb lesions (TaïNP and BFP) or ulcerative anogenital skin lesions (BFP, NKNP, and LMNP)Citation9.
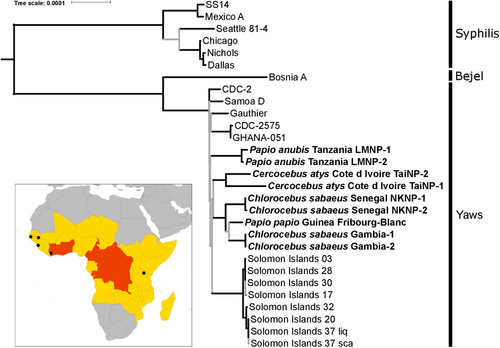
NHP-infecting Treponema pallidum strains are indicated in bold. In this maximum likelihood tree, nodes with less than 95% bootstrap approximation support are indicated with gray lines. Tip labels indicate the NHP species sampled, the country of origin, and the sample ID. The scale is in nucleotide substitutions per site. The inset is a map of Africa showing the sites of origin of NHP samples from which a TP genome was determined (indicated with black circles). The 2013 yaws status of countries, based on the World Health Organization’s Global Health Observatory (http://www.who.int/gho/en/), are indicated by color: gray indicates no previous history of yaws infections in humans, yellow indicates a country previously endemic for yaws for which the current status is unknown, and red indicates countries that are currently considered endemic for yaws
Using a PCR-based assay, we demonstrated the presence of TP in skin lesion biopsies or swabs from NHPs inhabiting TaïNP (C. atys), BFP, and NKNP (C. sabaeus). TP infection in olive baboons (P. anubis) from LMNP had previously been confirmedCitation6. Two samples per NHP population were selected for whole-genome sequencing based on a high TP copy number or the ability to amplify long PCR fragments (Supplementary Table S2). To overcome the presence of background host genomic DNA, we used targeted DNA capture coupled with next generation sequencing to reconstruct whole TP genomesCitation2,Citation8. Following quality filtering, removal of PCR duplicates, merging of different sequencing runs from the same sample, and mapping against the TPE strain Fribourg-Blanc reference genome, we obtained a range of 22,886–470,303 DNA sequencing reads per sample. All samples showed at least an 80% coverage of the reference genome with a depth coverage of three or higher; the average genome coverage depth was between 6.1-fold and 121.0-fold (Supplementary Table S3).
We generated maximum likelihood, Bayesian and maximum parsimony trees based on the genomes reconstructed in our study and all available reference genomes (total sequence length: 1,133,379 nucleotides). In all trees, the TPE and TPA strains formed reciprocally monophyletic groups, with a mean TPE/TPA strain divergence of 0.099%. NHP-infecting TP strains all clustered with human-infecting TPE strains (Fig. ; Supplementary Figure S1). The TPE clade exhibited a star-like branching pattern with basal branches that were very short and received low statistical support. Importantly, this pattern does not support a clear reciprocal monophyly of the TPE strains infecting humans and NHPs. In line with this result, the minimum divergence between strains infecting humans and NHPs was lower than the maximum divergence among human or NHP-infecting strains (0.011% versus 0.015% and 0.024%). The human-infecting TPE strains Samoa D, CDC-2, CDC-2575, Ghana-051, and Gauthier, which span a broad geographic and temporal range (at least four decades), were less divergent from each other than the two strains infecting sooty mangabeys from a single social group at TaïNP, which were collected in the same week (0.011% versus 0.017% sequence divergence, respectively). While intra-group strain divergence was low for the two African green monkey populations and the olive baboons (0.0003% and 0.0017%, respectively), intra-species strain divergence among African green monkeys was relatively high compared to the divergence observed between the two most divergent human strains (0.0094% versus 0.015%).
We determined the complete genome sequence and structure for the TPE strain from sample LMNP-1 (average depth of coverage: ×169; GenBank: CP021113; Supplementary Table S5-6)Citation12. The genome structure of the LMNP-1 strain was the same as those of published complete genomes of human-infecting TPE strains and that of the simian strain Fribourg-Blanc. Furthermore, the genome of the LMNP-1 strain was more similar to that of the human-infecting TPE Gauthier strain than the simian isolate Fribourg-Blanc, showing differences at 266 and 325 chromosomal positions, respectively. Most differences were single-nucleotide substitutions or small indels (Supplementary Table S7). The LMNP-1 and Gauthier strains exhibited the same number of 24-bp repeats in the TP_0470 gene (n = 25), and the Gauthier strain had only one 60-bp repeat more than the LMNP-1 strain in the arp gene (LMNP-1 n = 9 vs. Gauthier n = 10). All 60-bp repeats in the arp gene of the LMNP-1 strain were of Type II and were identical to other TPE strainsCitation13. The tprK gene of the LMNP-1 strain had only three variable regions, V5–V7, compared to other TPE strains. In addition to differences in the TP_0433, TP_0470, and tprK genes, relatively large indels were identified in TPEGAU_0136 (33-nt long deletion; specific for the strains Gauthier and Samoa D), TPFB_0548 (42-nt long deletion; specific for strain Fribourg-Blanc), and TPEGAU_0858 (79-nt long deletion; specific for strain Gauthier), and in the intergenic regions (IGRs) between TPEGAU_0628 and TPEGAU_0629 (302-nt long deletion; specific for strain Gauthier) and TPFB_0696 and TPFB_0697 (430-nt long insertion; specific for strain Fribourg-Blanc); the lengths of the other sequence differences ranged between 1 and 15 nt. The structures of the rRNA operons in the LMNP-1 genome (coordinates 231,180–236,139; 279,584–284,533; according to TPE strain Gauthier: NC_016843.1) were similar to those in strains Gauthier, CDC-2, and Fribourg-Blanc, but were different than those in strains Samoa D, Samoa F, and CDC-1. The LMNP-1 16S–5S–23S region was identical in both operons, and the 23S rRNA sequences were identical to those in other TPE strains except for strain Fribourg-Blanc (having a single-nucleotide difference at position 458). We did not observe any mutations associated with macrolide resistance (e.g., A2058G, A2059G)Citation14. When the two NHP-infecting TPE strains (Fribourg-Blanc and LMNP-1) were compared to the closest human-pathogenic TPE strains (CDC-2 and Gauthier) only 7.2 and 9.1% of all coding sequences (77 and 97 coding sequences out of 1065) contained amino acid substitutions, respectively, suggesting limited functional divergence among these strains (Supplementary Table S7-9).
Our findings unambiguously indicate that at least three African NHP species (representing four populations) from West and East Africa currently suffer from treponematosis caused by TPE. Taking into account the isolation of the Fribourg-Blanc strain from Guinea baboons in 1966 and its recent sequencing and identification as a member of the TPE cladeCitation12, there are currently four African NHP species and five populations whose symptoms can be explained by TPE infections. Coupled with a growing number of clinical and serological observationsCitation6,Citation7,Citation9,Citation10, these findings suggest that infection of NHPs with TPE is common throughout sub-Saharan Africa. Thus, humans are not the exclusive host for the yaws bacterium, as NHPs are infected with the same bacterial agent.
TPE strains in NHPs exhibit considerable genetic diversity, which at least equals that found among published human-infecting TPE strains. Importantly, we found no evidence for a clear sub-differentiation of NHP-infecting and human-infecting TPE strains, i.e., these strains did not form well-supported reciprocally monophyletic groups. Rather, the star-like topology of our phylogenomic tree suggests a rapid initial radiation of the ancestor of TPE, which may have involved transmission across primate species barriers in the relatively distant past (with respect to the TPE clade depth). These results neither support nor allow us to exclude a possible recent transmission of TPE between NHPs and humans, especially due to the large geographic and temporal separation between the two groups of samples compared in this study. A major hurdle in identifying such potential transmission events is the availability of bacterial genomes. Despite large numbers of human cases, very few genomes have been determined from human-infecting TPE strains and only from a very limited geographic range. Generating additional human-infecting TPE genomes represents an important area of research, the results of which, when coupled with the genomes of the NHP-infecting TPE strains presented here, could enable the detection of recent zoonotic transmission events, should any exist.
Since yaws has not been reported for several decades in humans in countries where we observed NHPs to be infected with TPE, we expect that if transmission of TPE between NHPs and humans occurs, it does so at a very low frequency (as is the case for many zoonotic diseases). Of course, such a low frequency of zoonotic transmission would not alone explain the reemergence of yaws, which is largely (or entirely) the consequence of continued human-to-human transmission. However, now that eradication of yaws appears within reachCitation15, the finding that TPE strains circulate in NHPs certainly supports the call for more research into their diversity and zoonotic potential.
Supplementary Information
Download MS Word (131.8 KB)Supplementary Information
Download MS Word (15.6 KB)Supplementary Information
Download MS Word (15.6 KB)Supplementary Information
Download MS Word (19.2 KB)Supplementary Information
Download MS Word (13.9 KB)Supplementary Information
Download MS Word (137.1 KB)Supplementary Information
Download MS Word (15.4 KB)Supplementary Information
Download MS Word (14.3 KB)Supplementary Information
Download MS Word (24.2 KB)Supplementary Information
Download MS Word (29.3 KB)Supplementary Information
Download MS Word (57.6 KB)Acknowledgements
This article represents a chapter in the doctoral dissertation of J.F.G. and has benefited greatly from the input of his supervisor committee and advisors Jonathan Davies, David Marcogliese, Charles Nunn, and Louis Lefebvre. Simone Lüert and Ulla Thiesen are thanked for discussions and technical support. We thank the Ivorian Ministry of Environment and Forests, the Ministry of Research, the directorship of the TaïNP, the Office Ivoirien des Parcs et Réserves, the Centre Suisse de Recherche Scientifique, the Taï Chimpanzee Project and the Taï Monkey project and their teams of field assistants for their support. We thank Jonathan Müller-Tiburtius, Therese Löhrich, Sylvain Lemoine, Simon T. Kannieu, Daniel Gnimion, Richard Peho, and Martina Magris for field assistance at TNP. For assistance and support in BFP, we thank the Department of Parks and Wildlife Management and the Forestry Department. For assistance and support with sequencing and read processing at the RKI, we are grateful to Wojciech Dabrowski, Julia Hinzmann, Andreas Nitsche, and Julia Tesch. We would like to thank the Senegalese authorities, in particular the officials of NKNP, who helped our work in Senegal immensely. We also thank Annick Abeille, who performed the initial PCR screening and sequencing of the Senegalese green monkey samples. For the work on East African simian strains, we would like to thank the TAWIRI, TANAPA, LMNP staff, especially the chief park warden, the ecology monitoring unit and park rangers, and the Tanzania Commission for Science and Technology (COSTECH). Details about the funding that supported this project are presented in the Supplementary Materials.
Data availability
All raw sequence read files have been deposited in NCBI as part of the BioProject PRJNA343706.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41426-018-0156-4).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Research Data
References
- GiacaniLLukehartSAThe endemic treponematosesClin. Microbiol. Rev.201427 89 11510.1128/CMR.00070-13
- AroraNOrigin of modern syphilis and emergence of a pandemic Treponema pallidum clusterNat. Microbiol.201621624510.1038/nmicrobiol.2016.245
- MarksMYaws: towards the WHO eradication targetTrans. R Soc. Trop. Med. Hyg.201611031932010.1093/trstmh/trw032
- World Health Organization. Eradication of yaws: procedures for verification and certification of interruption of transmission (World Health Organization, Geneva, 2018).
- AsieduKFitzpatrickCJanninJEradication of yaws: historical efforts and achieving WHO’s 2020 targetPLoS Negl. Trop. Dis.20148e301610.1371/journal.pntd.0003016
- FelsenfeldOWolfRHSerological reactions with treponemal antigens in nonhuman primates and the natural history of treponematosis in manFolia Primatol.19711629430510.1159/000155411
- Fribourg-BlancAMollaretHHNatural treponematosis of the African primatePrimates Med.19693113121
- GogartenJFTools for opening new chapters in the book of Treponema pallidum evolutionary historyClin. Microbiol. Infect.20162291692110.1016/j.cmi.2016.07.027
- KnaufSTreponema infection associated with genital ulceration in wild baboonsVet. Pathol.20124929230310.1177/0300985811402839
- ChumaISWidespread Treponema pallidum infection in nonhuman primates, TanzaniaEmerg. Infect. Dis.2018241002100910.3201/eid2406.180037
- PetrosovaHWhole genome sequence of Treponema pallidum ssp. pallidum, strain Mexico A, suggests recombination between yaws and syphilis strainsPLoS Negl. Trop. Dis.20126e183210.1371/journal.pntd.0001832
- ZobanikovaMWhole genome sequence of the Treponema Fribourg-Blanc: unspecified simian isolate is highly similar to the yaws subspeciesPLoS Negl. Trop. Dis.20137e217210.1371/journal.pntd.0002172
- HarperKNThe sequence of the acidic repeat protein (arp) gene differentiates venereal from nonvenereal Treponema pallidum subspecies, and the gene has evolved under strong positive selection in the subspecies that causes syphilisFEMS Immunol. Med. Microbiol.20085332233210.1111/j.1574-695X.2008.00427.x
- LukehartSAMacrolide resistance in Treponema pallidum in the United States and IrelandN. Engl. J. Med.200435115415810.1056/NEJMoa040216
- MarksMMathematical modeling of programmatic requirements for yaws eradicationEmerg. Infect. Dis.201723222810.3201/eid2301.160487