
Abstract
Background
Usher syndrome (USH) typically leads to deaf-blindness, requiring the provision of extensive education and rehabilitation services. Therefore, investigating the prevalence is crucial to requests for proper government support for USH patients.
Objective
The aim was to perform a nationwide epidemiologic survey of USH in Japan to estimate the prevalence of USH and reveal the relative frequency and characteristics of the three USH subtypes.
Methods
To estimate the number of USH patients visiting hospitals over a 1-year period, 1,628 hospitals were randomly selected from all Departments of Otorhinolaryngology and Ophthalmology in Japan. Subsequently, we collected data regarding the clinical characteristics of each patient treated and the results of genetic testing, if performed.
Results
We found that the prevalence of USH was at least 0.4 per 100,000 population. The frequency of clinical subtypes and causal genes for USH were consistent with previous reports. Also, we demonstrated the feasibility of genetic counseling for USH patients based on the results of genetic testing.
Conclusion
USH is a rare disease, but requires social support due to the severity of symptoms. To minimize these issues, understanding the clinical characteristics and performing comprehensive genetic testing could allow early and accurate diagnosis as well as medical intervention.
Chinese abstract
背景:Usher 综合征 (USH) 通常会导致耳聋-失明, 需要得到教育和康复服务的提供。因此, 为 USH 患者请求政府提供适当的支持, 调查发病率至关重要。
目的:在日本进行全国性USH流行病学调查, 以估计USH 的患病率, 并揭示三种 USH 类型的相对频率和特征。
方法:为了估计 1 年期间就诊的 USH 患者数量, 从日本所有耳鼻喉科和眼科随机选择了 1,628 家医院。随后, 收集了每位接受治疗的患者的临床特征资料以及基因检测的结果(如果进行了检测)。
结果:我们发现 USH 患病率至少为每 100,000 人中 0.4 人。USH 的临床亚型的频率和致病基因与之前的报道一致。此外, 我们还根据基因检测结果证明了对 USH 患者进行遗传咨询的可行性。
结论:USH 是一种罕见疾病, 但由于症状的严重性, 需要社会支持。为减少这些问题, 了解临床特征并进行全面的基因检测可以使得早期准确的诊断以及医疗干预成为可能。
Introduction
Usher syndrome (USH) is an autosomal recessive disorder characterized by hearing loss (HL), retinitis pigmentosa (RP) and vestibular dysfunction. Three clinical subtypes can be distinguished. USH type 1 (USH1) is the most severe among the three subtypes because of profound HL, absent vestibular responses, and prepubertal onset RP. USH type 2 (USH2) is characterized by congenital moderate-to-severe HL, with a high-frequency sloping configuration. The vestibular function is normal and onset of RP is in the first or second decade. The onset of the visual symptoms, such as night blindness, in USH usually occurs several years later than that in USH1. USH type 3 (USH3) is characterized by variable onset of progressive HL, variable onset of RP, and variable impairment of vestibular function (normal to absent) [Citation1]. USH typically leads to combined neurosensory deficits of the auditory and visual systems that are not only potentially devastating to the lives of the persons affected, but may also prevent independent living, thus requiring the provision of extensive education and rehabilitation services. Therefore, investigating the prevalence of USH is crucial to requests for proper government support for USH patients; however, most studies to date have been performed in Europe and the US, not in Asian countries.
USH is genetically heterogeneous, and 9 genes have been reported to be associated with USH, based on the Hereditary Hearing Loss Homepage (https://hereditaryhearingloss.org/). The disease-causing genes for USH1 include MYO7A (USH1B), USH1C (USH1C), CDH23 (USH1D), PCDH15 (USH1F), and USH1G (USH1G). Mutations in USH2A (USH2A), ADGRV1 (USH2C), and WHRN (USH2D) are responsible for USH2, and the only gene responsible for USH3 is CLRN1 (USH3A). Some of the above USH genes are also responsible for non-syndromic HL or RP. CDH23 and PCDH15 are responsible for both nonsyndromic deafness and USH1 (USH1D and USH1F, respectively). For these two genes, a genotype-phenotype correlation has been reported, and Usher syndrome is known to result from homozygosity for truncating nonsense, frameshift and splice site mutations [Citation2,Citation3]. On the other hand, missense mutations typically cause non-syndromic deafness (DFNB12 and DFNB23, respectively). Additionally, mutations in USH2A have resulted in USH2A as well as nonsyndromic RP [Citation4]. We have reported the mutation spectrum in Japanese USH1 patients [Citation5], but the gene frequencies in Japanese USH2 and USH3 remain unclear.
The diagnosis of USH in childhood, based on clinical phenotypes, can be challenging as patients often appear to have non-syndromic HL only in their youth until RP develops in later years. However, early diagnosis through genetic testing allows the diagnosis of USH prior to the appearance of visual symptoms as well as appropriate genetic counseling [Citation6,Citation7].
Herein, we performed a nationwide epidemiologic survey of USH in Japan to estimate the prevalence of USH. We reported the relative frequencies and characteristics of the three USH subtypes. Further, based on the USH patients reported in the survey, we showed how genetic counseling can be useful in the lives of USH patients through a USH case series.
Materials and methods
The nationwide epidemiologic survey
This nationwide epidemiologic survey was undertaken according to the previously established protocols proposed by the Research Committee on the Epidemiology of Intractable Diseases in Japan [Citation8]. The overall method has been previously described [Citation9]. The survey comprised first and second queries. The aim of the first query was to estimate the number of USH patients visiting hospitals over the 1-year period from 1 January 2017 through 31 December 2017, whereas the aim of the second was to elucidate the patients’ clinical characteristics.
The targets of the first query were selected from all Departments of Otorhinolaryngology and Ophthalmology that deal with USH in Japan by stratified random sampling according to the number of hospital beds. The sampling proportions were as follows: general hospitals with ≤99, 100–199, 200–299, 300–399, 400–499, and 500+ hospital beds were 5, 10, 20, 40, 80, and 100%, respectively; university hospitals were 100%. In addition, for specialized hospitals in which many USH patients would have received medical treatment, the sampling proportion was 100%. Selected departments were asked to complete a mail-back questionnaire on the annual numbers of USH patients who visited the department in the preceding year. We sent reminders to non-respondents by post at 2 and 3 months after the initial mailing.
If a department responded that it had at least one patient in the first query, we sent a second query. The second query was designed to collect data regarding the clinical characteristics of each patient treated and consisted of the following items: clinical USH subtype (type 1–3), sex, date of birth, date of symptom onset, family history of USH, and results of genetic testing, if performed.
The survey was approved by the Ethics Committee of Shinshu University School of Medicine (Approval number: 4314).
Statistical analysis
In consideration of the sampling and response proportions in the first query, we estimated the total number of USH patients in Japan according to the following formula: the estimated total number of patients = reported number of patients/(sampling proportion × response proportion). The period prevalence for the year was calculated based on the Japanese population on 1 October 2017.
Results
At total of 1,628 of the 4,147 departments in Japan were sampled as the survey targets of the first query (sampling rate, 39.3%) of which 1,161 responded (response rate, 71.3%). We received reports for 313 USH patients. Based on these data, we estimated the total number of USH patients in Japan to be 510. The prevalence per 100,000 population was 0.4 ().
Table 1. Prevalence of Usher syndrome by country.
The characteristics of the USH patients from the second query are shown in and and . Of the 133 USH patients for whom data were available, 24.8% (n = 33) were diagnosed with USH1, 35.3% (n = 47) with USH2, and 24.8% (n = 33) with USH3 (). Among the three USH subtypes, patients without a family history were most common. Regarding both HL and RP, the mean ages at symptom onset and diagnosis were earlier for patients with USH1 than for patients with USH2 and USH3. Additionally, the mean onset age of the HL in all subtypes of USH was earlier than the mean onset age of RP (). Of 80 USH patients who received genetic testing, causal genes were identified in 35 patients (43.8%).
Figure 1. Frequency of causal genes in USH1 and USH2. USH1: Usher syndrome type 1, USH2: Usher syndrome type 2.
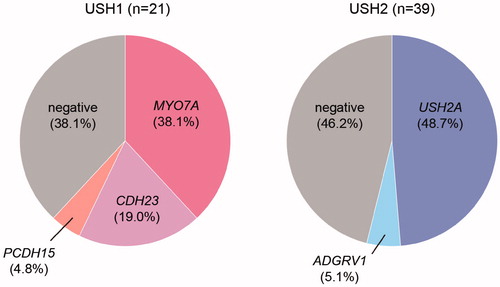
Table 2. Characteristics of Usher syndrome patients reported in this study.
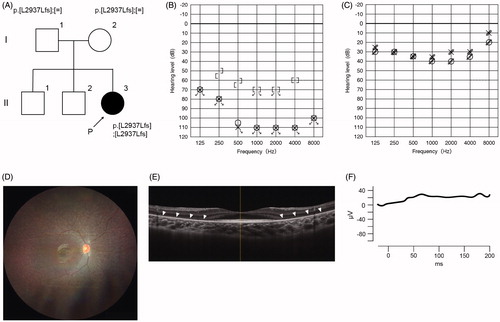
Of the 21 USH1 patients, USH1B (MYO7A) was the most common, accounting for 38.1%, followed by USH1D (CDH23) and USH1F (PCDH15) in 19.0% and 4.8%, respectively (). Case #13 is worth describing in detail. This 9-year-old girl had not passed the newborn hearing screening bilaterally, and subsequently visited an ENT clinic where she was diagnosed with bilateral profound HL (). She was able to hold her head up at 7 months old and walk at 2 years. She received sequential bilateral cochlear implantation for the right ear at the age of 1 year 10 months and for the left ear at the age of 3. At the age of 5, genetic testing revealed pathogenic variants in the CDH23 gene (p.[L29378Lfs];[L29378Lfs]) (). Hearing thresholds with bilateral CI were excellent (). To distinguish between non-syndromic HL and USH1, we recommended that an ophthalmologist be consulted. Although the proband showed no visual symptoms at that time, a fundus examination of both eyes revealed atrophic changes in the peripheral retina and attenuated retinal vessels (). Also, there was a decrease in the thickness of the outer retinal layers on spectral-domain optic coherence tomography (SD-OCT), ). The recordings of full field electroretinograms (ERGs) showed non-recordable patterns bilaterally (). Therefore, the proband was diagnosed with RP. On the basis of the results of hearing ability, genetic testing, and ophthalmologic evalutations, the patient was then diagnosed with USH1D. We provided genetic counseling to the family about (1) the risk of future constriction of the visual field and vision loss and (2) the benefits of bilateral CI. After the counseling, she became aware of night blindness at the age of 6. At present, she is simply nearsighted.
Figure 2. Clinical findings of Case #13. (A) Pedigree and genetic testing results for the proband and family. (B) The audiogram of the proband indicated bilateral profound hearing loss. (C) Hearing thresholds with bilateral cochlear implants were adequate. (D) Fundus examination revealed atrophic peripheral retina and attenuated retinal vessels (E) SD-OCT demonstrated that the thickness of outer retinal layers was decreased. Arrowheads indicate thinned outer retinal layers. (F) Full-field ERG of rod- and cone-mixed maximum response showed non-recordable patterns bilaterally.
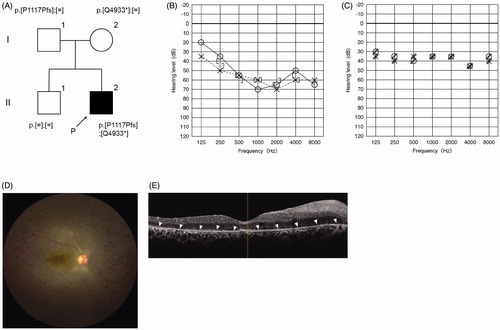
Of the 39 USH2 patients, USH2A (USH2A) was the most frequent (48.7%), followed by USH2C (ADGRV1) (5.1%) (). Case #8, a 22-year-old man, was diagnosed with HL and started to wear hearing aids at the age of 3. At the age of 13, he was aware of night blindness and was subsequently diagnosed with RP based on the results of some ophthalmologic tests. A fundus examination revealed retinal bone-spicule pigmentation, macular atrophy, and attenuated retinal vessels (). Also, SD-OCT indicated that the thickness of outer retinal layers was decreased (). Thereafter, genetic analysis revealed that he carried two compound heterozygous pathogenic variants (p.[P1117Pfs];[Q4933*]) in the USH2A gene (). At the age of 17, we provided genetic counseling to the proband and parents about (1) the possibility of nonprogression in hearing loss and (2) the risk of future constriction of the visual field and vision loss. At present, he has high-frequency HL, and aided thresholds are adequate (). Although the loss of visual field is already severe, his hearing ability could remain stable for at least 6 years. He worked as a hairdresser, but he decided to think about future work based on the natural course of HL and RP in USH2 based on the results of the genetic counseling.
Figure 3. Clinical findings of Case #8. (A) Pedigree and genetic testing results for the proband and family. (B) The audiogram of the proband indicated bilateral high-frequency hearing loss. (C) Hearing thresholds with bilateral hearing aids were good. (D) Fundus examination revealed retinal bone-spicule pigmentation, macular atrophy, and attenuated retinal vessels. (E) SD-OCT indicated that the thickness of outer retinal layers was decreased (arrowheads).
Discussion
There had been no epidemiological research on USH for approximately 20 years prior to the present study, which revealed that the overall prevalence in Japan was 0.4 per 100,000 population, a much lower prevalence than those in previous studies in Europe and the US () [Citation10,Citation11, Citation13–15]. One explanation for this difference could be the subject recruitment area. In addition, except for the research in Denmark [Citation13], the data in other studies were collected from specified cities where a high-volume center existed, suggesting that the prevalence in these studies might have been over-estimated. A second explanation could be patient age. Rosenberg et al. studied USH patients who were aged between 20 and 49 years old [Citation13]. This criterion was reasonable, because younger patients may have incomplete symptoms owing to late presentation of symptomatic retinitis pigmentosa in children and as older patients may not visit a hospital routinely due to severe deaf-blindness. However, this study was based on all age groups in the Japanese population, which can cause difficulties in the estimation of the true proportion. Taken together, an estimated prevalence of 0.4/100000 should be considered as an absolute minimum figure. Additionally, finding genetically distinct subtypes (like the Finnish over-representation of Usher syndrome type 3 [Citation12]) may finally lead to the detection of clusters causing regional epidemiological differences [Citation13].
The distribution among phenotypes varies from report to report; however, a 2:3 ratio between USH1 and USH2 was documented by Rosenberg et al. [Citation13], which is consistent with our results (USH1 24.8% vs. USH2 35.3%). Both USH1 and USH2 predominantly show congenital HL, but our findings indicated that the age at symptom onset and diagnosis for USH1 was younger than that for USH2. We think that this characteristic was affected by the severity of HL, but the newborn hearing screening could allow earlier diagnosis of HL for USH2. Also, comprehensive genetic testing could identify pathogenic variants in USH-responsible genes in children with nonsyndromic HL without visual symptoms, suggesting that they take ophthalmologic examinations, which could lead to the earlier diagnosis of USH1 and USH2, as in Case #13 in this study [Citation6,Citation7].
In this study, the diagnostic rate for USH1 by genetic testing was similar that in our previous report [Citation5], with the MYO7A gene most frequently implicated, followed by CDH23 and PCDH15. We did not find any USH1 caused by USH1C or USH1G. With regard to USH2, mutations in USH2A were the most frequent, followed by ADGRV1, which is consistent with the results of other reports [Citation16].
USH is predominantly diagnosed by clinical symptoms, but like Case #13, genetic testing allowed ‘early’ diagnosis prior to the appearance of visual symptoms and led to appropriate intervention. In cases in which USH1 patients did not receive CI, we were able to counsel them about advantages in terms of hearing abilities and social life following bilateral CI. In addition, approximately 120 deafness genes have been identified for non-syndromic hearing loss (https://hereditaryhearingloss.org/), and more than 60 RP genes have been identified for non-syndromic RP (https://sph.uth.edu/retnet/sum-dis.htm). There is a possibility that patients have unrelated pathogenic variants associated with both ‘non-syndromic hearing loss’ and ‘non-syndromic RP,’ leading to incorrect diagnoses. To address this issue, comprehensive genetic testing should be used to allow ‘accurate’ diagnosis. As in Case #8, the genetic analysis of USH could provide the opportunity for patients to consider their future.
This work is the first nationwide survey of USH in an Asian country, highlighting that the prevalence of USH was at least 0.4 per 100,000 population. The frequency of clinical subtypes and causal genes for USH were consistent with the findings of previous reports. Additionally, we demonstrated the feasibility of genetic counseling for USH patients based on the results of genetic testing.
In conclusion, based on the prevalence data, USH is a rare disease. However, it requires social support due to the severity of symptoms. To minimize these issues, understanding the clinical characteristics and performing comprehensive genetic testing could allow early and accurate diagnosis as well as medical intervention.
Acknowledgments
We would like to thank the participants of the Interactable Hearing Disorder Consortium: Dr. Katada Akihiro (Asahikawa Medical University), Drs. Noriko Ogasawara and Tomoko Shintani (Sapporo Medical University), Drs. Yumiko Kobayashi and Hiroaki Sato (Iwate Medical University), Dr. Seiji Kakehata (Yamagata University), Dr. Daisuke Kikuchi (Fukushima Medical University), Dr. Tetsuo Ikezono (Saitama Medical University), Dr. Kotaro Ishikawa (National Rehabilitation Center), Dr. Kyoko Shirai (Tokyo Medical University), Drs. Masahiro Takahashi and Satoshi Iwasaki (International University of Health and Welfare), Dr. Yasuhiro Arai (Yokohama City University), Dr. Hajime Sano (Kitasato Univerisity), Dr. Mayuri Okami (Tokai University), Dr. Hiroshi Nakanishi (Hamamatsu University School of Medicine), Dr. Tomoko Esaki (Aichi Children's Health and Medical Center), Dr. Michihiko Sone (Nagoya Univerisity), Dr. Jun Nakayama (Shiga University of Medical Science), Dr. Takayuki Okano (Kyoto University), Dr. Yumi Ota (Osaka University), Dr. Hiroshi Nishimura (National Hospital Organization Osaka National Hospital), Dr. Yasushi Naito (Kobe City Medical Center General Hospital), Drs. Yuko Kataoka, Akiko Sugaya and Yukihide Maeda (Okayama University), Dr. Shin Masuda (Hiroshima Prefectual Hospital), Drs. Kazuma Sugawara and Hiroshi Yamashita (Yamaguchi University), Dr. Naoto Hato (Ehime University), Dr. Takashi Nakagawa (Kyusyu University), Drs. Akira Ganaha and Tetsuya Tono (Miyazaki University), Dr. Ikuyo Miyanohara (Kagoshima University), Dr. Mikio Suzuki (Ryukyu University).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Yan D, Liu XZ. Genetics and pathological mechanisms of Usher syndrome. J Hum Genet. 2010;55(6):327–335.
- Ahmed ZM, Riazuddin S, Ahmad J, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003;12(24):3215–3223.
- Astuto LM, Bork JM, Weston MD, et al. CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 2002;71(2):262–275.
- Rivolta C, Sweklo EA, Berson EL, et al. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66(6):1975–1978.
- Yoshimura H, Iwasaki S, Nishio SY, et al. Massively parallel DNA sequencing facilitates diagnosis of patients with Usher syndrome type 1. PLoS One. 2014;9(3):e90688.
- Moteki H, Yoshimura H, Azaiez H, et al. USH2 caused by GPR98 mutation diagnosed by massively parallel sequencing in advance of the occurrence of visual symptoms. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):123S–128S.
- Yoshimura H, Iwasaki S, Kanda Y, et al. An Usher syndrome type 1 patient diagnosed before the appearance of visual symptoms by MYO7A mutation analysis. Int J Pediatr Otorhinolaryngol. 2013;77(2):298–302.
- Nakamura Y, the Study Group on Epidemiologic Research for Instractable Diseases. A manual of a nationwide epidemiological survey for estimating the number of patients and assessing clinico-epidemiological characteristics of patients with intractable diseases (3rd edition) [in Japanese]. 2017. Available at: http://www.jichi.ac.jp/dph/nanbyou/manual_2017.pdf (Accessed 12 August, 2021).
- Ohfuji S, Furuichi Y, Akahoshi T, et al. Japanese periodical nationwide epidemiologic survey of aberrant portal hemodynamics. Hepatol Res. 2019;49(8):890–901.
- Boughman JA, Vernon M, Shaver KA. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis. 1983;36(8):595–603.
- Grondahl J. Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway. Clin Genet. 1987;31(4):255–264.
- Pakarinen L, Karjalainen S, Simola KO, et al. Usher’s syndrome type 3 in Finland. Laryngoscope. 1995;105(6):613–617.
- Rosenberg T, Haim M, Hauch AM, et al. The prevalence of usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet. 1997;51(5):314–321.
- Hope CI, Bundey S, Proops D, et al. Usher syndrome in the city of Birmingham-prevalence and clinical classification. Br J Ophthalmol. 1997;81(1):46–53.
- Spandau UH, Rohrschneider K. Prevalence and geographical distribution of Usher syndrome in Germany. Graefes Arch Clin Exp Ophthalmol. 2002;240(6):495–498.
- Jouret G, Poirsier C, Spodenkiewicz M, et al. Genetics of Usher syndrome: new insights from a Meta-analysis. Otol Neurotol. 2019;40(1):121–129.