
Abstract
Cytochrome P450 (CYP) enzymes are heme-containing monooxygenases that catalyze metabolisms of various endogenous and exogenous compounds. They constitute a superfamily of enzymes present in various organisms including mammals, plants, bacteria, and insects. CYPs are diverse and metabolize a wide variety of substrates, but their structures are largely conserved. In this study, bioinformatic analyses of CYP enzymes were performed in seed plants and other organisms’ protein sequences, including 39 species of 19 different families. According to the conserved motifs obtained by MEME and MAST tools, four motifs were common in most seed plants. The structural and functional analyses of six selected species of seed plants and other organisms were investigated by ProtParam, SOPMA, Predotar 1.03, SignalP 4.1 and TMHMM 2.0 tools in ExPASy database. The tertiary structures of Arabidopsis thaliana as a sample of seed plants and five samples of other organisms were predicted by Phyre2 server using “2hi4.1.A” model (PDB accession code: 2hi4). The protein sequences were aligned with ClustalW algorithm by MegAlign (DNAStar Lasergene 12.1) and MEGA 6.06, then the phylogenetic tree was constructed using the neighbor-joining (NJ) method. In protein–protein interactions analysis by STRING 9.1 tool, six enriched pathways were identified in Arabidopsis thaliana and other species. According to the results, there is a high similarity among CYP proteins of different species in seed plants and other organisms, so they should be derived from a common ancestor. The obtained data provide a background for bioinformatic studies of the function and evolution of these proteins in seed plants.
Abbreviations:
- CO: Carbon monoxide
- CYP: Cytochrome P450
- ER: Endoplasmic reticulum
- GRAVY: Grand average of hydropathicity
- KEGG: Kyoto Encyclopedia of Genes and Genomes
- MAST: Motif Alignment and Search Tool
- MEGA: Molecular Evolutionary Genetic Analysis
- MEME: Multiple Em for Motif Elicitation
- NCBI: National Center for Biotechnology Information
- PDB: Protein Data Bank
- RMSD: Root mean square deviation
- SOPMA: Self-Optimized Prediction Method with Alignment
- STRING: Search Tool for the Retrieval of Interacting Genes/Proteins
Introduction
Cytochromes P450 (CYPs), which are called hemoproteins, exist in different organisms including mammals, plants, bacteria and insects. In eukaryotes, P450 is membrane-bound and its function, which is inserting one molecule of oxygen into its substrate with its heme prosthetic group, plays a role in substrate oxidation. This catalytic reaction requires a pair of electrons transferred from NADPH through the NADPH-cytochrome P450 reductase enzyme, a P450 redox partner, to target P450s (Ortiz de Montellano Citation2005) (Figure ). There are many isoforms of CYPs, categorized into 18 families and 57 subfamilies based on their amino acid identity, and named on the basis of the overall amino acid sequence (Nelson Citation2011). During the processes of CYP evolution, a pseudogene may be generated which can be identified by either its aberrant coding region or transcriptional silence or both (Wilde Citation1985, Nebert et al. Citation1988).
Figure 1. The cytochrome P450 catalytic cycle.
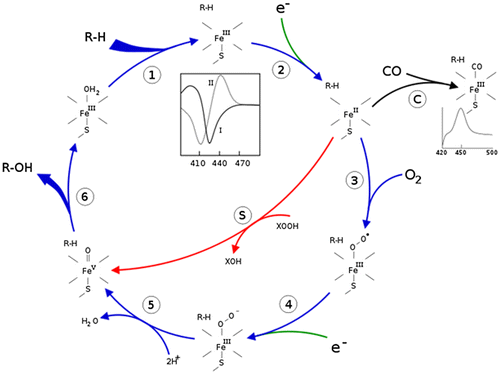
CYP enzymes have been recognized in all organisms: plants, animals, fungi, protists, bacteria, archaea, and viruses (Lamb et al. Citation2009). The most common reaction of CYPs is a monooxygenase reaction, e.g. insertion of one atom of oxygen into the aliphatic position of an organic substrate (RH) while the other oxygen atom is reduced to water:
RH + O2 + NADPH + H+ → ROH + H2O + NADP+
The active site of CYP contains a heme iron center. The iron is tethered to the P450 protein via a thiolate ligand derived from a cysteine residue. This cysteine and several flanking residues are highly conserved in known CYPs. They are the key enzymes involved in drug metabolism, accounting for about 75% of total metabolism (Guengerich Citation2007). They deactivate most drugs, either directly or by facilitated excretion from the body. Also, many substances are bio-activated by CYPs to form their active compounds. Many drugs may increase or decrease the activity of several CYP isozymes by enzyme induction (inducing the biosynthesis of an isozyme) or enzyme inhibition (inhibiting the activity of the CYP). Naturally occurring compounds may also induce or inhibit CYP activity. For example, bioactive compounds such as bergamottin, dihydroxybergamottin, and paradicin-A which are found in grapefruit juice and some other fruit juices inhibit CYP3A4-mediated metabolism of certain medications, leading to increased bioavailability and, thus, the strong possibility of overdosing (Bailey and Dresser Citation2004).
CYPs have been extensively examined in human, mice, rats, dogs, and less so in zebrafish, in order to facilitate use of these model organisms in drug discovery and toxicology. Recently they have also been found in avian species, in particular turkeys, which may be a good model for cancer research in humans (Rawal et al. Citation2010). CYP1A5 and CYP3A37 in turkeys were found to be very similar to the human CYP1A2 and CYP3A4, respectively, in terms of their kinetic properties as well as in the metabolism of aflatoxin B1 (Rawal and Coulombe Jr Citation2011).
In insects, CYPs are membrane-bound enzymes that play important roles in endogenous metabolisms (i.e. metabolisms of steroid molting, juvenile hormones, and pheromones) and xenobiotic metabolisms, as well as detoxification of insecticides (Feyereisen Citation1999). For example, CYP6G1 is linked to insecticide resistance in DDT-resistant Drosophila melanogaster (McCart Citation2008), and CYP6Z1 in the mosquito malaria vector, Anopheles gambiae, metabolizes DDT (Chiu et al. Citation2008). CYP genes of the CYP6 family have been reported in several insects to be involved in metabolism of plant defensive chemicals (Scott et al. Citation1998, Kahn and Durst Citation2000) and insecticides (Feyereisen Citation1999, Scott Citation1999, Scott and Wen Citation2001).
Plant CYPs are involved in a wide range of biosynthetic reactions, leading to various fatty acid conjugates, plant hormones, defensive compounds, or medically crucial drugs. Terpenoids, which represent the main class of characterized natural plant compounds, are often substrates for plant CYPs. There are several potential CYP inducers present in wheat. They include cyclic hydroxamic acids and related benzoxazolinones, a crucial group of allelochemicals present in Gramineae members (wheat, rye, maize, etc.). These hydroxamic acids have been shown to be involved in plant defense against fungi, bacteria, and insects (Frey et al. Citation1997, Fomsgaard et al. Citation2004). CYP6B members have not only been described to respond to a wide range of allelochemicals present in host plants (indole-3-carbinol, jasmonate, xanthotoxin, etc.) but also to synthetic chemicals, such as insecticides (Li et al. Citation2002a, Citation2002b). CYPs may have an important role in establishing compatible interactions and host plant resistance. The mode of resistance offered by the host plant against hessian fly (Mayetiola destructor) is governed predominantly by single genes which are completely to partially dominant (Zantoko and Shukle Citation1997).
Eukaryotic CYPs generally range in length from approximately 480 to 560 amino acids. All members of this superfamily share a common globular to triangular structural framework which consists of a relatively alpha-helix rich carboxy-terminal half and relatively beta-sheet rich amino-terminal half. Most of the beta-sheets and alpha helices are laid in roughly the same plane as the prosthetic heme group (Danielson Citation2002).
The significant reactivity and substrate promiscuity of CYPs have long attracted the attention of chemists (Chefson and Auclair Citation2006). Recent progress towards realizing the potential of using CYPs towards difficult oxidations have included: (1) eliminating the need for natural co-factors by replacing them with inexpensive peroxide containing molecules (Chefson et al. Citation2006); (2) exploring the compatibility of CYPs with organic solvents (Chefson and Auclair Citation2007); and (3) the use of small, non-chiral auxiliaries to predictably direct CYP oxidation.
In this study, we performed bioinformatic analyses on this enzyme in seed plants and other samples of organisms for prediction of different structures. Our results could provide bioinformatics background for the investigation of CYPs in seed plants.
Material and methods
Database search and sequence retrieval
A total of 39 CYP protein sequences of 19 different families in seed plants and other organisms were collected from National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov).
Conserved motifs analyses
The motifs of protein sequences were found using the program of Multiple Em for Motif Elicitation (MEME; version 4.9.1) (Bailey et al. Citation2009) and Motif Alignment and Search Tool (MAST; version 4.9.1) (Bailey and Gribskov Citation1998) at http://meme.nbcr.net/meme. The parameters of MEME analyses were applied as follows: distribution of motif occurrences, zero or one per sequence; number of different motifs, 10; minimum motif width, six; and maximum motif width, 50.
Structural and functional analyses
Several online web services and software were used for analyses of CYP proteins in seed plants and other organisms. Comparative and bioinformatic analyses were carried out online at the website ExPASy (http://expasy.org/tools). Physicochemical parameters of CYP proteins were analyzed by ProtParam (http://web.expasy.org/protparam) (Gasteiger et al. Citation2005). The secondary structure prediction was analyzed by SOPMA (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) (Geourjon and Deleage Citation1995).
Prediction of mitochondrial and plastid targeting sequences was performed by the Predotar 1.03 server (https://urgi.versailles.inra.fr/predotar/predotar.html) and prediction of signal peptide cleavage sites was carried out by SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP) (Petersen et al. Citation2011). The identification of transmembrane helices in proteins was analyzed by TMHMM 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/) (Moller et al. Citation2001).
Tertiary structure prediction
The tertiary structure prediction analysis of six samples of CYP proteins was performed by the Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) (Kelley and Sternberg Citation2009) using profile–profile matching and secondary structure. WebLab ViewerLite 4.2 was used for 3D structure visualization. The TM-score server (http://zhanglab.ccmb.med.umich.edu/TM-score/) (Zhang and Skolnick Citation2004, Xu and Zhang Citation2010) was used to find the backbone similarities and differences of obtained models. Also, they were superposed on each other by the SuperPose web server (http://wishart.biology.ualberta.ca/SuperPose/) (Maiti et al. Citation2004).
Multiple sequence alignment and phylogenetic analysis
The multiple sequence alignment of CYP proteins was performed with ClustalW algorithm implemented in MegAlign (DNAStar Lasergene 12.1) (http://www.dnastar.com/) and Molecular Evolutionary Genetic Analysis (MEGA 6.06) (http://www.megasoftware.net) (Tamura et al. Citation2013) with default parameters. The phylogenetic tree was constructed using the neighbor-joining (NJ) method and the bootstrap test carried out with 1000 replicates.
Protein–protein interactions
The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING 9.1) database (http://string-db.org/) was used to predict the interacting proteins (Franceschini et al. Citation2013). The database contains information from numerous sources, including experimental repositories, computational prediction methods and public text collections.
Results
Identification and characterization of CYP proteins
In the present study, 34 CYP protein sequences of 14 different families in seed plants including Amborellaceae, Arecaceae, Brassicaceae, Cucurbitaceae, Euphorbiaceae, Fabaceae, Malvaceae, Musaceae, Poaceae, Rosaceae, Rutaceae, Salicaceae, Solanaceae, and Vitaceae were retrieved from NCBI with FASTA format. In order to compare the results, five different species of other organisms including Caenorhabditis elegans (Rhabditidae), Danio rerio (Cyprinidae), Drosophila melanogaster (Drosophilidae), Homo sapiens (Hominidae) and Mus musculus (Muridae) were also selected and added to seed plant sequences whose characteristics including scientific name, type, family, abbreviation, accession number, and length of amino acid sequence were listed in Table .
Table 1. List of 39 different species of seed plants and other organisms in this study.
Identification of conserved motifs
The analyses of conserved motifs of CYP proteins were performed by MEME and MAST programs. A conserved motif is a sequence pattern that occurs repeatedly in a group of related protein sequences. MEME represents motifs as position-dependent letter-probability matrices which describe the probability of each possible letter at each position in the pattern and motifs in MAST are represented as position-dependent scoring matrices that describe the score of each possible letter at each position in the pattern. Based on the results, 10 conserved motifs of CYPs were found (Table , Figure ). As can be seen in Figure , motifs 1, 3, 4, and 7 were common in 35 species.
Table 2. The conserved motifs found by the MEME tool in 39 species of seed plants and other organisms.
Figure 2. The conserved motifs for CYP proteins in seed plants and other organisms found using the MEME tool. The motifs are shown as different-colored boxes.
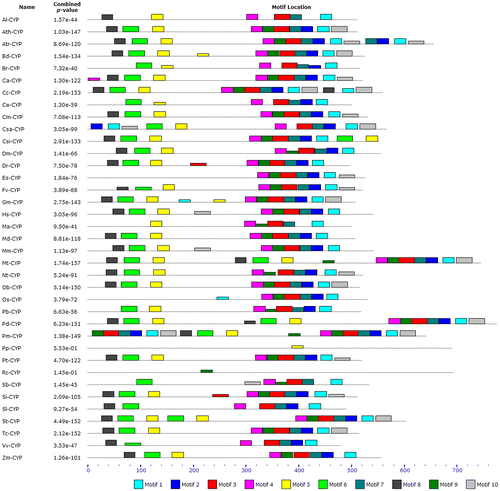
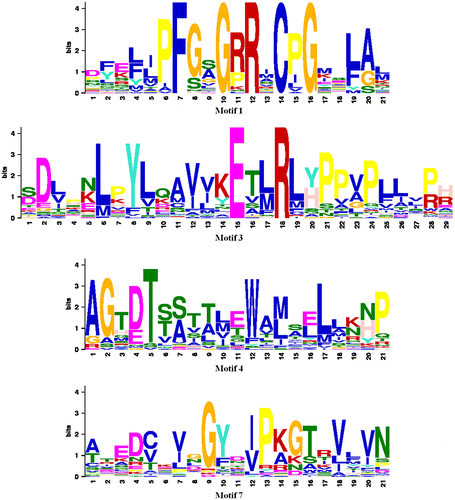
Figure 3. Sequence-specific MEME conserved motifs for CYP proteins.
Structural and functional predictions of CYP proteins
In order to investigate the structures and functions of CYP proteins, Arabidopsis thaliana (thale cress) as a representative of seed plants and five different samples of other organisms including Caenorhabditis elegans (roundworm), Danio rerio (zebrafish), Drosophila melanogaster (fruit fly), Homo sapiens (human), and Mus musculus (house mouse) were selected and analyzed by bioinformatics tools. The primary structure of selected CYPs was analyzed by ProtParam server. ProtParam computes various physicochemical properties that can be deduced from a protein sequence including the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity (GRAVY) (Table ). Secondary structure prediction was performed by the SOPMA (Self-Optimized Prediction Method with Alignment) server. This server calculates the percent of alpha-helix, extended strand, beta-sheet and random coil in protein sequences. According to the results, most parts of the CYPs are alpha helix and random coil (Table ).
Table 3. The results of primary structure analysis and secondary structure prediction in Ath-CYP and other samples of organisms.
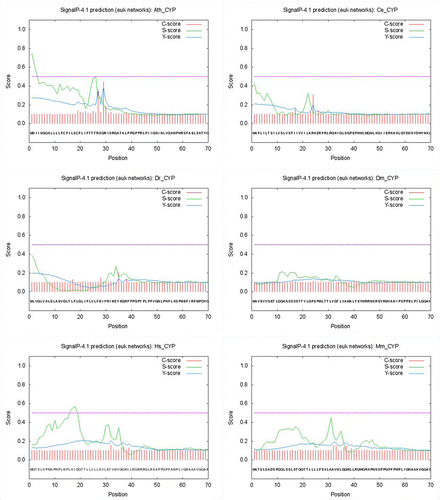
Post-translational modification predictions were analyzed by Predotar 1.03 and SignalP 4.1 servers. Predotar recognizes the N-terminal targeting sequences of classically targeted precursor proteins. For each protein sequence, Predotar provides a probability estimate as to whether the sequence contains a mitochondrial, plastid or ER targeting sequence. The fourth number (elsewhere) is simply the estimated probability that no targeting sequence is present. These estimates assume that the sequence in question was randomly chosen from a proteome in which about 10% of proteins are targeted to mitochondria, 10% to plastids and 20% to the ER. Based on the results of Predotar server, only three CYP proteins including Ath-CYP, Ce-CYP and Dr-CYP contained ER targeting sequence and the rest of the protein samples had no targeting sequence (Table ). The SignalP 4.1 server predicts the presence and location of signal peptide cleavage sites in protein sequences. The method incorporates a prediction of cleavage sites and a signal peptide/non-signal peptide prediction based on a combination of several artificial neural networks. A signal peptide is a short (5–30 amino acids long) peptide present at the N-terminus of the majority of newly synthesized proteins that are destined towards the secretory pathway. According to the results of SignalP analysis, there was no signal peptide in CYP proteins in six sample organisms (Table , Figure ).
Table 4. The results of post-translational modification and topology predictions by different tools in Ath-CYP and other samples of organisms.
Figure 4. Prediction of signal peptide cleavage sites in Ath-CYP and other samples of organisms by the SignalP 4.1 server. C-score (raw cleavage site score), S-score (signal peptide score), and Y-score (combined cleavage site score) are shown.
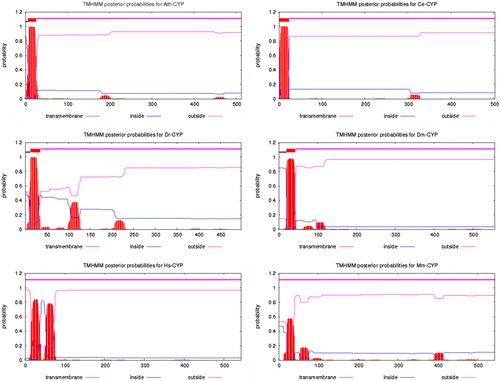
Topology prediction was carried out by the TMHMM 2.0 server. This server is for prediction of transmembrane helices in proteins. Transmembrane helices are visible in structures of membrane proteins determined by X-ray diffraction. They may also be predicted on the basis of hydrophobicity scales. Because the interior of the bilayer and the interiors of most proteins of known structure are hydrophobic, it is presumed to be a requirement of the amino acids that span a membrane that they be hydrophobic as well. However, membrane pumps and ion channels also contain numerous charged and polar residues within the generally non-polar transmembrane segments. According to the results of the TMHMM 2.0 server, one transmembrane helix can be seen in Ce-CYP and Dr-CYP but there were not any of them in Hs-CYP and Mm-CYP (Table and Figure ).
Figure 5. Prediction of transmembrane helices in Ath-CYP and other samples of organisms by the TMHMM 2.0 server.
Tertiary structure prediction
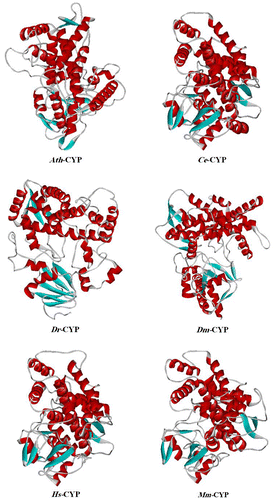
The tertiary structure prediction of Ath-CYP, Ce-CYP, Dr-CYP, Dm-CYP, Hs-CYP, and Mm-CYP was performed by the Phyre2 (Protein Homology/analogY Recognition Engine) server. It uses the alignment of hidden Markov models via HHsearch (Söding Citation2005) to significantly improve the accuracy of alignment and detection rate. It also incorporates a new ab initio folding simulation called Poing (Jefferys et al. Citation2010) to model regions of the proteins with no detectable homology to known structures. In this analysis, the models of Ath-CYP and other samples were predicted using “2hi4.1.A” model (PDB accession code: 2hi4), with 100% confidence (Figure ).
Figure 6. Tertiary structure prediction of CYP protein in Arabidopsis thaliana and other samples of organisms, established by the Phyre2 server using “2hi4.1.A” model (PDB accession code: 2hi4). The α-helix is shown helix-shaped in red, the beta sheet wide ribbon-shaped in blue, and the random coil line-shaped in gray. The tertiary structure is shown as a solid ribbon model by WebLab ViewerLite 4.2.
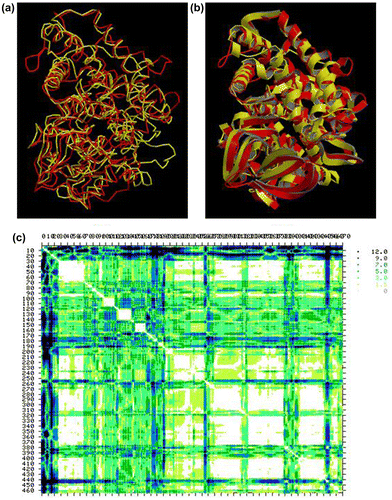
Furthermore, the structural similarities and differences between Ath-CYP and five other models were analyzed using the TM-score server. TM-score is a metric for measuring the structural similarity of two protein models. It is designed to solve two major problems in the traditional metrics such as RMSD (root mean square deviation): (1) TM-score measures the global fold similarity and is less sensitive to the local structural variations; (2) the magnitude of TM-score for random structure pairs is length-independent. TM-score has the value (0,1], where 1 indicates a perfect match between two structures. Following strict statistics of structures in the PDB, scores below 0.17 correspond to randomly chosen unrelated proteins whereas scores higher than 0.5 assume generally the same fold (http://zhanglab.ccmb.med.umich.edu/TM-score/). According to the obtained results, the highest similarity of five sample species to Arabidopsis thaliana belonged to Caenorhabditis elegans with TM-score = 0.6676. Because tertiary structure is actually much more conserved than sequence, its comparisons allow us to look even further back into biological prehistory (Maiti et al. Citation2004). The most common method for tertiary structure comparison is called structure superposition (or superimposition). So, in order to further evaluate Ath-CYP and Ce-CYP models, superposition of their structures was carried out by the SuperPose web server (Figure ).
Figure 7. Superposition of Ath-CYP and Ce-CYP models by the SuperPose web server. (a) Backbone and (b) ribbon superposed structures. The Ath-CYP is shown in red and Ce-CYP in yellow. (c) Difference distance matrix. The lighter and darker regions indicate more similar and different structures, respectively. The default display for SuperPose’s difference distance plot shows six graded cutoffs. Differences between 0 and 1.5 A° are white, differences between 1.5 and 3.0 A° are yellow, differences between 3.0 and 5.0 A° are a light green, differences between 5 and 7 A° are colored dark turquoise, differences between 7 and 9 A° are colored dark blue and those greater than 9 are colored black.
Multiple sequence alignment and phylogenetic analysis
The multiple sequence alignment of CYP proteins was performed with ClustalW algorithm implemented in MegAlign (DNAStar Lasergene 12.1) and (MEGA 6.06) with default parameters. The percentage identities of Ath-CYP and five samples of other organisms with seed plants were shown in Table . According to the results obtained by MegAlign, the maximum and minimum identity of Ath-CYP belonged to Capsella rubella (3.1%) and Eutrema salsugineum (42.8%), respectively.
Table 5. The percentage identity of Ath-CYP and other samples of organisms with seed plants in the multiple sequence alignment by MegAlign (DNAStar Lasergene 12.1).
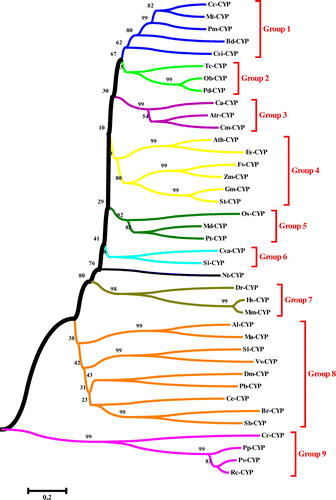
The phylogenetic tree was constructed using the neighbor-joining (NJ) method and the bootstrap test carried out with 1000 replicates by MEGA 6.06 software (Figure ). Based on the phylogenetic relationships, CYPs were divided into nine groups, designated from Group 1 to Group 9.
Figure 8. Phylogenetic tree of CYP proteins from 39 different species in seed plants and other organisms, using the ClustalW method (MEGA 6.06). The neighbor-joining (NJ) method was used to construct the tree. The percentage of 1000 bootstrap replicates is given at each node. Based on the phylogenetic tree result, 39 protein sequences of CYP were defined, approximating nine major groups marked with different colors.
Functional interaction network analysis of CYP proteins
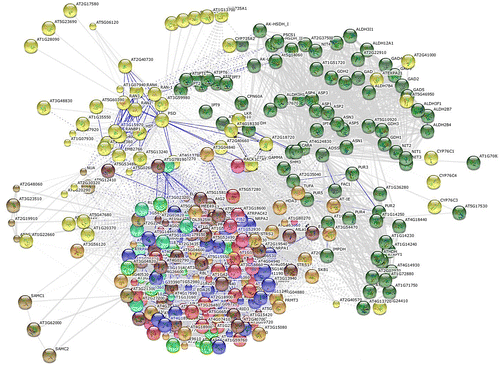
In order to predict the interacting proteins, Ath-CYP was mapped to the STRING 9.1 tool as a sample of seed plants. STRING is a database of known and predicted protein interactions. The interactions include direct (physical) and indirect (functional) associations. According to the results, 10 functional partners and six enriched pathways of KEGG were identified in the network analysis (Table and Figure ).
Table 6. Characteristics of input protein (Ath-CYP), predicted functional partners and KEGG pathways in STRING 9.1 tool.
Figure 9. Interactive network view of predicted CYP proteins using the STRING 9.1 tool.
Discussion
CYP enzymes are a diverse and ancient class of enzymes that date back to three billion years ago, and play a key role in plant, animal, and microbial biology (Nebert and Russell Citation2002). They participate in oxidation, peroxidation and reduction of compounds ranging from pharmaceutical drugs to environmental chemicals to endogenous bioactive molecules (Rendic and Di Carlo Citation1997). These proteins, named for the absorption band at 450 nm of their carbon-monoxide bound form, are one of the largest super families of enzyme proteins. The CYP genes are found in the genomes of virtually all organisms, but their number has vastly increased in plants.
In the present study, we used bioinformatics methods to investigate the characteristics of CYP in 39 species of 19 different families of seed plants and other organisms. At first, the MEME and MAST analyses of CYP proteins were performed in order to find patterns of conserved motifs. As can be seen in Table and Figures and , among 10 conserved motifs in 39 different species, motif 1 (21 aa, E-value = 3.7e-284), motif 3 (29 aa, E-value = 3.7e-322), motif 4 (21 aa, E-value = 1.0e-215), and motif 7 (21 aa, E-value = 2.9e-161) were common in 35 species. According to these results, there was no common conserved motif in all the species, also two CYPs including Cr-CYP and Pv-CYP had no common conserved motif with any protein of other species.
In primary structure analysis, sequence length, molecular weight, theoretical isoelectric point (pI value), total number of negatively (Asp+Glu) and positively (Arg+Lys) charged residues, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) were computed (Table ). In the study of six CYP proteins consisted of Ath-CYP (512 aa), Ce-CYP (502 aa), Dr-CYP (498 aa), Dm-CYP (556 aa), Hs-CYP (543 aa), and Mm-CYP (543 aa), it was found that Dm-CYP and Ath-CYP had the highest and lowest molecular weight, respectively. Moreover, the computed isoelectric point for CYPs was between 6.45 and 9.18. Isoelectric point is a pH at which a protein carries no net charge. It is of significance in protein purification as it is the pH at which solubility is often minimal and mobility in an electrofocusing system is zero. The largest number of negatively charged residues belonged to Ce-CYP and Dm-CYP (68), whereas the lowest was observed in Mm-CYP (49). Also, Ce-CYP and Dr-CYP had the highest number of positively charged residues (69) and the lowest one (53), respectively. Computed values of instability index of CYPs were between 37.13 (Ce-CYP) and 42.99 (Ath-CYP). A protein whose instability index is smaller than 40 is predicted as stable, a value above 40 predicts that the protein may be unstable. The aliphatic index of a protein is defined as the relative volume occupied by aliphatic side chains (alanine, valine, isoleucine, and leucine). It may be regarded as a positive factor for the increase of thermostability of globular proteins. As it was shown in Table , like the instability index, the highest aliphatic index belonged to Ath-CYP (99.59), while the lowest was observed in Ce-CYP (85.84). As a result, high aliphatic index in CYPs indicated structural stability. In the obtained values of GRAVY, only negative values were detected. The GRAVY value for a protein is calculated as the sum of hydropathy values of all the amino acids, divided by the number of residues in the sequence.
In secondary structure analysis of six selected species, alpha helix was present at the highest percentage (46.78–49.80%), and beta turn at the lowest (3.39–4.42%) (Table ). According to these results in six representative species, it can be assumed that the results would be the same in the species.
Two servers including Predotar 1.03 and SignalP 4.1 were used in post-translational modification prediction (Table ). Based on the results of the Predotar server, three representative species including Dm-CYP, Hs-CYP, and Mm-CYP were not predicted to be targeted to mitochondria. Furthermore, none of the six samples were expected to be targeted to plastid, and only three CYP proteins including Ath-CYP, Ce-CYP, and Dr-CYP contained endoplasmic reticulum targeting sequence. In the signal peptide analysis, it was shown that none of six selected species had it, so CYP is not a secretory protein (Figure ).
Transmembrane domains of CYPs were studied in topology prediction (Table , Figure ). According to the results, Ath-CYP, Ce-CYP, Dr-CYP, and Dm-CYP had no transmembrane domains; however, there were also none in Hs-CYP and Mm-CYP. So, the number, presence or absence of transmembrane domains is not predictable in different species.
The 3D structure is the ultimate goal of protein structure prediction and it is necessary to fully understand protein function. As can be seen in Figure by ExPASy’s Phyre2 server, it was found that six sample CYPs have an intricate spatial architecture which is a very similar crystal structure to human microsomal P450 1a2, whose catalytic activity has been determined (Sansen et al. Citation2007). According to the model, chain A contains “cytochrome P450 1a2” and there are three types of non-polymeric entities, namely: 2-phenyl-4 h-benzo[h]chromen-4-one, protoporphyrin IX containing Fe, and water. Also, the values of TM-score were computed between Ath-CYP model and the others. According to the results of TM-score server, the estimated values of Ce-CYP, Dr-CYP, Dm-CYP, Hs-CYP, and Mm-CYP were 0.6676, 0.4110, 0.2031, 0.2023, and 0.2002, respectively. As a result, the highest and lowest structural similarity of Arabidopsis thaliana to five other models belonged to Caenorhabditis elegans and Mus musculus.
The multiple sequence alignment of CYP proteins by MegAlign showed the percentage identify of Ath-CYP and five CYP samples of other species. As can be seen in Table , except for Dr-CYP and Hs-CYP, the CYPs had lowest identity to Cr-CYP (Capsella rubella P450 protein), though the highest identities were different. In phylogenetic analysis among 39 CYP proteins, they were categorized in nine main groups. Group 8, with nine members, was the largest group in CTP phylogeny, and Group 6, with two members, was the smallest. Nt-CYP located outside of any groups. Ath-CYP located in Group 4, which made a subgroup with Es-CYP (bootstrap 99). Three samples of other CYPs, including Dr-CYP, Hs-CYP, and Mm-CYP, formed Group 7 without any species of seed plants. Finally, Dm-CYP and Ce-CYP located in Group 8 with high distance to seed plants. According to the phylogeny tree, CYPs were derived from an ancestor and evolved into different groups.
Genes involved in related biological pathways are usually expressed cooperatively for their functions, and thus information on their interaction is key to understanding the biological systems at the molecular level (Eisen et al. Citation1998). To further explore which genes are possibly regulated by CYP protein or pathway, a protein–protein interaction network was assembled (Table , Figure ). For this reason, Arabidopsis thaliana was selected as the representative of seed plants and examined by the STRING 9.1 tool. Based on Table , 10 functional partners were identified. Partner CYP735A1 with the highest score (0.932) had functions including “electron carrier”, “heme binding”, “iron ion binding”, “monooxygenase”, and “oxygen binding”, while partners AT5G15810 and AT3G56330 with the lowest score (0.513) had two functions: “RNA binding”, and “tRNA (guanine-N2-)-methyltransferase activity”. Other obtained partners in this analysis had scores between 0.924 and 0.513, and had functions including “adenylosuccinate synthase”, “expansin-like protein”, “nucleobase, nucleoside, nucleotide and nucleic acid transmembrane transporter”, and “adenylate dimethylallyltransferase”. Additionally, six pathways of KEGG, including “zeatin biosynthesis”, “metabolic pathways”, “biosynthesis of secondary metabolites”, “RNA transport”, “purine metabolism”, and “alanine, aspartate and glutamate metabolism”, were identified in the network analysis.
Bioinformatics can play a vital role in the analysis and interpretation of genomic and proteomic data. It uses methods and technologies from mathematics, statistics, computer sciences, physics, biology, and medicine (Romano et al. Citation2011). It can be a powerful tool for predicting the function of a protein from its amino acid sequence and has revolutionized the studies of organisms’ metabolism (Darabi et al. Citation2012, Darabi and Farhadi-Nejad Citation2013, Seddigh and Darabi Citation2014, Darabi and Seddigh Citation2015, Seddigh and Darabi Citation2016). In this research, bioinformatic analyses of CYPs in seed plants and other organisms exhibited similarities of these proteins in different families and the obtained data provide a background for bioinformatic studies of the function and evolution of the proteins in seed plants.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Bailey DG, Dresser GK. 2004. Interactions between grapefruit juice and cardiovascular drugs. Am J Cardiovasc Drugs. 4(5):281–297.10.2165/00129784-200404050-00002
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009). “MEME SUITE: tools for motif discovery and searching”. Nucleic Acids Res 37 (Web Server issue): W202–208.
- Bailey TL, Gribskov M. 1998. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 14(1):48–54.10.1093/bioinformatics/14.1.48
- Chefson A, Auclair K. 2006. Progress towards the easier use of P450 enzymes. Molecular BioSystems. 2(10):462–469.10.1039/b607001a
- Chefson A, Auclair K. 2007. CYP3A4 activity in the presence of organic cosolvents, ionic liquids, or water-immiscible organic solvents. ChemBioChem. 8(10):1189–1197.10.1002/(ISSN)1439-7633
- Chefson A, Zhao J, Auclair K. 2006. Replacement of natural cofactors by selected hydrogen peroxide donors or organic peroxides results in improved activity for CYP3A4 and CYP2D6. ChemBioChem. 7(6):916–919.10.1002/cbic.v7:6
- Chiu TL, Wen Z, Rupasinghe SG, Schuler MA. 2008. Comparative molecular modeling of Anopheles gambiae CYP6Z1, a mosquito P450 capable of metabolizing DDT. Proc. Natl. Acad. Sci. 105(26):8855–8860.10.1073/pnas.0709249105
- P.B. Danielson Bentham Science Publisher. 2002. The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr Drug Metab. 3(6):561–597.10.2174/1389200023337054
- Darabi M, Farhadi-Nejad H. 2013. Study of the 3-hydroxy-3-methylglotaryl-coenzyme A reductase (HMGR) protein in Rosaceae by bioinformatics tools. Caryologia. 66(4):351–359.10.1080/00087114.2013.856089
- Darabi M, Masoudi-Nejad A, Nemat-Zadeh G. 2012. Bioinformatics study of the 3-hydroxy-3-methylglotaryl-coenzyme A reductase (HMGR) gene in Gramineae. Mol Biol Rep. 39(9):8925–8935.10.1007/s11033-012-1761-2
- Darabi M, Seddigh S. 2015. Bioinformatic characterization of aspartic protease (AP) enzyme in seed plants. Plant Syst Evol. 301(10):2399–2417.10.1007/s00606-015-1236-8
- Eisen MB, Spellman PT, Brown PO, Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Nat. Acad. Sci. 95(25):14863–14868.10.1073/pnas.95.25.14863
- Feyereisen R. 1999. Insect P450 enzymes. Annu Rev Entomol. 44(1):507–533.10.1146/annurev.ento.44.1.507
- Fomsgaard IS, Mortensen AG, Carlsen SC. 2004. Microbial transformation products of benzoxazolinone and benzoxazinone allelochemicals—a review. Chemosphere. 54(8):1025–1038.10.1016/j.chemosphere.2003.09.044
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C. 2013. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41(D1):D808–D815.10.1093/nar/gks1094
- Frey M, Chomet P, Glawischnig E, Stettner C, Grün S, Winklmair A, Eisenreich W, Bacher A, Meeley RB, Briggs SP. 1997. Analysis of a chemical plant defense mechanism in grasses. Science. 277(5326):696–699.10.1126/science.277.5326.696
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. 2005. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics protocols handbook. Humana Press; p. 571–607.
- Geourjon C, Deleage G. 1995. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci: CABIOS. 11(6):681–684.
- Guengerich FP. 2007. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 21(1):70–83.
- Jefferys BR, Kelley LA, Sternberg MJ. 2010. Protein folding requires crowd control in a simulated cell. J Mol Biol. 397(5):1329–1338.10.1016/j.jmb.2010.01.074
- Kahn R, Durst F. 2000. Function and evolution of plant cytochrome P450. Recent Adv Phytochem. 34:151–189.10.1016/S0079-9920(00)80007-6
- Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols. 4(3):363–371.10.1038/nprot.2009.2
- Lamb DC, Lei L, Warrilow AG, Lepesheva GI, Mullins JG, Waterman MR, Kelly SL. 2009. The first virally encoded cytochrome P450. Journal of Virology. 83(16):8266–8269.10.1128/JVI.00289-09
- Li X, Berenbaum MR, Schuler MA. 2002a. Plant allelochemicals differentially regulate Helicoverpa zea cytochrome P450 genes. Insect Mol Biol. 11(4):343–351.10.1046/j.1365-2583.2002.00341.x
- Li X, Schuler MA, Berenbaum MR. 2002b. Jasmonate and salicylate induce expression of herbivore cytochrome P450 genes. Nature. 419(6908):712–715.10.1038/nature01003
- Maiti R, Van Domselaar GH, Zhang H, Wishart DS. 2004. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 32(Web Server):W590–W594.10.1093/nar/gkh477
- McCart C. 2008. Dissecting the insecticide-resistance-associated cytochrome P450 gene Cyp6g1. Pest Manage Sci. 64(6):639–645.10.1002/(ISSN)1526-4998
- Moller S, Croning MD, Apweiler R. 2001. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 17(7):646–653.10.1093/bioinformatics/17.7.646
- Nebert D, Jones J, Owens J, Puga A. 1988. Evolution of the P450 gene superfamily. Prog Clin Biol Res. 274:557–576.
- Nebert DW, Russell DW. 2002. Clinical importance of the cytochromes P450. The Lancet. 360(9340):1155–1162.10.1016/S0140-6736(02)11203-7
- Nelson DR (2011). Progress in tracing the evolutionary paths of cytochrome P450. Biochim. Biophys. Acta, Proteins and Proteomics. 1814(1): 14–18.10.1016/j.bbapap.2010.08.008
- Ortiz de Montellano PR (2005). Cytochrome P450: structure, mechanism, and biochemistry. New York: Kluwer Academic/Plenum Piblishers, ISBN 0-306-48324-6.
- Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 8(10):785–786.10.1038/nmeth.1701
- Rawal S, Coulombe RA Jr. 2011. Metabolism of aflatoxin B1 in Turkey liver microsomes: The relative roles of cytochromes P450 1A5 and 3A37. Toxicol Appl Pharmacol. 254(3):349–354.10.1016/j.taap.2011.05.010
- Rawal S, Kim JE, Coulombe R Jr. 2010. Aflatoxin B1 in poultry: Toxicology, metabolism and prevention. Res Vet Sci. 89(3):325–331.10.1016/j.rvsc.2010.04.011
- Rendic S, Carlo F. 1997. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev. 29:413–580.10.3109/03602539709037591
- Romano P, Giugno R, Pulvirenti A. 2011. Tools and collaborative environments for bioinformatics research. Briefings in Bioinformatics. 12(6):549–561.10.1093/bib/bbr055
- Sansen S, Yano JK, Reynald RL, Schoch GA, Griffin KJ, Stout CD, Johnson EF. 2007. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J Biol Chem. 282(19):14348–14355.10.1074/jbc.M611692200
- Scott JG. 1999. Cytochromes P450 and insecticide resistance. Insect Biochem Mol Biol. 29(9):757–777.10.1016/S0965-1748(99)00038-7
- Scott JG, Liu N, Wen Z. 1998. Insect cytochromes P450: diversity, insecticide resistance and tolerance to plant toxins. Comp. Biochem. Physiol. Part C: Pharmacol. Toxicol. 121(1):147–155.
- Scott JG, Wen Z. 2001. Cytochromes P450 of insects: the tip of the iceberg. Pest Manage Sci. 57(10):958–967.10.1002/(ISSN)1526-4998
- Seddigh S, Darabi M. 2014. Comprehensive analysis of beta-galactosidase protein in plants based on Arabidopsis thaliana. TURKISH J. Biol. 38(1):140–150.10.3906/biy-1307-14
- Seddigh S, Darabi M. 2016. Proteomics comparison of aspartic protease enzyme in insects. Turkish J. Biol. 40(1):69–83.
- Seddigh S, Darabi M. 2015. Structural and phylogenetic analysis of α-glucosidase protein in insects. Biologia. 70(6):812–825.
- Soding J. 2005. Protein homology detection by HMM–HMM comparison. Bioinformatics. 21(7):951–960.10.1093/bioinformatics/bti125
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.10.1093/molbev/mst197
- Wilde C. 1985. Pseudogenes. Crit. Rev. Biochem. 19(4):323–352.
- Xu J, Zhang Y. 2010. How significant is a protein structure similarity with TM-score= 0.5?. Bioinformatics. 26(7):889–895.10.1093/bioinformatics/btq066
- Zantoko L, Shukle R. 1997. Genetics of virulence in the Hessian fly to resistance gene H13 in wheat. J Hered. 88(2):120–123.10.1093/oxfordjournals.jhered.a023069
- Zhang Y, Skolnick J. 2004. Scoring function for automated assessment of protein structure template quality. Proteins: Struct. Funct., and Bioinf. 57(4):702–710.10.1002/(ISSN)1097-0134