
ABSTRACT
To improve our capabilities to model surrogate fuels, particularly in respect to polycyclic aromatic hydrocarbons (PAHs) and soot formation, the pyrolysis of n-heptane is studied in a single-pulse shock across a wide temperature range (900–1700 K) at 20 bar nominal pressure and 4 ms residence time. Three different initial fuel mole fractions are considered, 103, 502, and 2000 ppm of n-heptane in argon. Fuel and intermediate species, including aromatics up to three-ring structures, are measured using gas chromatography and mass spectrometry diagnostics. An ongoing detailed chemical kinetic model for PAH chemistry has been updated to successfully capture the fuel decomposition, the formation of small hydrocarbons, and the concentration of the main PAH products. Major reaction pathways to PAHs are highlighted as well as the role of important intermediate species.
Introduction
n-Heptane is widely used as a single-component surrogate for diesel fuel and as a key reference fuel for gasoline (Curran et al. Citation1998). Thus, its pyrolytic and oxidation chemistry has been studied for decades. At high temperatures, it dissociates into small hydrocarbon products, mainly C2, C3, and C4, which, in absence of oxygen or at fuel-rich conditions, may subsequently react to form larger aromatic species such as the polycyclic aromatic hydrocarbons (PAHs). In general, understanding PAH formation during combustion processes paves the way for the researchers to develop soot formation kinetic models, which can help them improve combustion efficiency and reduce emissions. Hence, a detailed investigation of n-heptane pyrolysis can reveal the fuel breakdown and subsequent molecular weight growth processes.
n-Heptane pyrolysis has long been pursued using numerous experimental configurations and methodologies that cover a wide range of pressure, temperature, and initial fuel concentration conditions. The list of literature investigations concerning n-heptane pyrolysis is reported in , and it includes only the studies performed in absence of oxygen as a detailed review on the oxidation chemistry of n-heptane is beyond the scope of the present work.
Table 1. Literature review on experimental investigations concerning n-heptane pyrolysis.
Among the reported investigations, only few articles provide detailed speciation measurements. The pioneering work by Appleby, Avery, and Meerbott (Citation1947) employed a flow technique to explore the thermal breakdown of n-heptane between 823 and 903 K at pressures of 1 and 8.7 atm. Murata and Saito (Citation1974, Citation1975) studied the decomposition of paraffins and 1-olefins, including n-heptane, at 973 K and 1 atm, and developed a molecular model to explain the product distribution. Both Bajus et al. (Citation1979), Pant and Kunzru (Citation1996), and Chakraborty and Kunzru (Citation2009) examined the steam cracking of n-heptane and determined the overall breakdown rate, by monitoring the formation of stable small hydrocarbon products using gas chromatography. Aribike and Susu (Citation1988, Citation1988) proposed a mechanistic model for n-heptane pyrolysis based on product distribution data acquired in a pulse reactor at atmospheric pressure and reaction durations of 0.40–1.02 s at 933–1053 K. Held and his coworkers (Citation1997) developed a simplified high-temperature pyrolysis and oxidation model for n-heptane that is validated against a variety of datasets, including their flow reactor experiments. More recent investigations have employed shock tube techniques for kinetic studies. Various time-history measurements were performed at Stanford University focusing on CH3 (Davidson, Oehlschlaeger, and Hanson Citation2007), C2H4 (Pilla, Davidson, and Hanson (Citation2011) and Pyun et al. (Citation2013)), and CH4 (Pyun et al. Citation2013) products. The single-pulse technique coupled to gas chromatography was also employed by Garner, Sivaramakrishnan, and Brzezinski (Citation2009) to investigate the pyrolysis of saturated and unsaturated C7 hydrocarbons, including n-heptane, in the temperature range 1000–1350 K at pressures of 25 to 50 atm with reaction times of 1–3 ms using a high-pressure shock tube. Yasunaga et al. (Citation2017, Citation2018) carried out a series of works to address the decomposition of 1% n-heptane in argon using a single-pulse shock tube at pressures of 1.0–2.5 atm and temperatures of 1000–1500 K for reaction durations ranging from 1.5 to 2.2 ms. Li et al. (Citation2021) conducted n-heptane and iso-octane pyrolysis experiments in a shock tube across a temperature range of 1200–2100 K and pressure range of 2.2–2.8 atm. Finally, among other techniques, Yuan et al. (Citation2011) reported speciation measurements in n-heptane pyrolysis at low pressure (400 Pa) and temperatures ranging from 780 to 1780 K using a flow reactor and synchrotron vacuum ultraviolet photoionization mass spectrometry.
According to the above review, past research has focused on the fuel decomposition reactivity and the formation of small hydrocarbons up to C6 intermediates. No studies have been conducted to investigate the formation of PAHs from n-heptane pyrolysis, though this latter focus is highly required for the development of clean combustion technologies. For this reason, the present work aims to provide shock tube speciation datasets spanning from small hydrocarbons to three-ring aromatics from n-heptane pyrolysis experiments at conditions relevant to modern combustion devices (20 bar, 900–1770 K). Our most recent kinetic model (Hamadi et al. Citation2022) has been updated to simulate and interpret the newly obtained experimental data. Kinetic insights into the formation of aromatic molecules from n-heptane pyrolysis will be addressed based on the experimental findings and kinetic modeling analyses.
Shock tube pyrolysis experiments
Pyrolysis experiments with three different n-heptane concentrations are performed using the high-purity single-pulse shock tube facility with dilute mole fractions of 103 ppm, 502 ppm, and 2000 ppm in bulk argon. A detailed description of the shock tube apparatus is well documented in our previous publications (Sun et al. Citation2022), so only a brief description is given here. The single-pulse shock tube apparatus consists of the driven (length: 6 m; inner diameter: 78 mm) and the driver (length: 3.7 m; inner diameter: 120 mm) sections, separated by a double diaphragm. The shock tube is equipped with a dump tank (volume 150 L) located near the diaphragm section, making it operate in a single pulse fashion. The low-pressure section is heated to 90°C to avoid condensation (or absorption) of fuel and reaction products. It is also pumped down to below 10−5 mbar with a turbo-molecular pump before each experiment. To avoid carbon deposits, the inner surface of the shock tube is cleaned regularly.
Four pressure sensors (CHIMIE METAL A25L05B) are mounted on the side wall of the driven section. The adjacent sensors are 150 mm apart, and the last sensor is 82 mm from the end wall. The distance and time difference between two successive pressure sensors are used to calculate the average incident shock velocity, which is then used to determine T5 and p5 behind the reflected shock wave by solving the conservation equations (Hugoniot Citation1887, Citation1889; Rankine Citation1870). The calculated T5 is subject to an uncertainty of ± 30 K due to the attenuation of the shockwave and the physical dimension of the pressure sensors (Hamadi et al. Citation2022). A PCB Piezotronics pressure sensor, shielded by a layer of room-temperature vulcanizing (RTV) silicone, is located on the end wall of the driven section to record the pressure history of each shock, from which the corresponding reaction time is determined (Tang and Brezinsky Citation2006). The reaction time for each experiment is defined as the time interval between the arrival of the incident shock wave and the time point at which the pressure drops to 80% p5 as a result of the quenching rarefaction waves. For the current experimental configuration, the nominal reaction time is 4 ms.
Stable species are withdrawn from the shock tube via an air-actuated valve and analyzed by the analytical system, which consists of three parts: an Agilent GC series 7890B, a Thermo Trace GC Ultra, and a Thermo DSQ mass spectrometer. The Agilent GC, designed to measure PAHs up to four rings, is equipped with a flame ionization detector (FID) coupled to a DB-17 ms column for heavy species separation, and a thermal conductivity detector coupled to a Molsieve 5A column for monitoring the absence of air and the dilution level by helium. An external valve box, coupled to the Agilent GC, diminishes the heavy compounds loss during sample storage and injection by maintaining the temperature at 320°C. The Thermo Trace GC is also equipped with an FID detector, connected to a HP Plot Q column for measuring light species up to mono-aromatics. The gas samples were also injected into the DSQ mass spectrometer, but in this case the MS analyses did not provide any additional information as all the heavy PAH compounds detected had been previously identified in our sequential studies on aromatic fuels.
The quantification of the measured species is based on the calibrations of the FID responses performed before the experiments. Standard gas mixtures are used for the calibration of the light species representing the C1-C5 hydrocarbons except for diacetylene (C4H2) and triacetylene (C6H2), whose calibration factors are obtained from high-temperature acetylene (C2H2) pyrolysis experiments through carbon atom conservation. Similarly, liquid fuels are used to calibrate benzene, heptane, toluene, styrene, and phenylacetylene. For all of the above calibrations, mixtures are prepared in stainless steel vessel using the partial pressure method. For small PAHs up to three rings, the calibration procedure differs. Here, the calibrations use gas-phase mixtures prepared in a heated glass vessel (200°C) (Sun et al. Citation2021b) to minimize surface adsorption (Comandini, Malewicki, and Brezinsky Citation2012). Uncertainties in species mole fractions are 5% for directly calibrated small species and 10% to 15% for PAH species calibrated in the gas phase. These uncertainties derive from the reproducibility of the single measure when repeated several times and the uncertainties in the composition of the initial reference calibration mixtures which reflect in the accuracy of the calibration curves.
The experiments are conducted with helium (Air Liquide, purity>99.995%) as the driver gas and dilute mixtures of n-heptane (Sigma – Aldrich, purity≥99.5%) in argon bath gas (Air Liquide, purity>99.9999%). The experimental gas mixtures are prepared in a 136 L electropolished stainless-steel cylinder that is evacuated in advance below 10−5 mbar with a turbomolecular pump. Before performing the experiments, the prepared gas mixtures are left overnight to homogenize, and the actual pre-shock compositions are analyzed by GC. The Supplementary Material provides all experimental results, consisting of the post-shock conditions (T5, p5), the measured pressure profiles, the defined reaction time, and the mole fraction measurements for individual species.
Kinetic modeling
The current work is a continuation of our previous serial work to build a kinetic model that can accurately predict the pyrolytic PAH formation under high-pressure and temperature conditions (the last version being published in (Hamadi et al. Citation2022)). The n-heptane sub-mechanism of Zhang et al. (Citation2016) is chosen as a substitute for the lumped heptane chemical mechanism. The n-heptane sub-mechanism involves unimolecular decomposition and H-abstraction reactions from the fuel and the corresponding fuel radicals. The reactions of cyclohexadiene with molecular hydrogen (CYC6H8+H) and subsequent fragmentation and the corresponding rate coefficients reported in (Wang, Villano, and Dean Citation2015) are incorporated into the current kinetic model. The rate coefficients of the unimolecular decomposition reaction of 1-pentene (C5H10-1) and 1-hexene (C6H12-1) are an update from the work of (Shao et al. Citation2020). Additionally, the rate coefficients of the H-abstraction reaction of ethylene (C2H4) are taken from the same LLNL PAH model (Shao et al. Citation2020).
Thermochemical data for most species are from the CRECK model (Pejpichestakul et al. Citation2019), as it is the basis of the current model (Sun et al. Citation2021c). For species introduced by reactions added or updated in this work, the thermochemical data are from the same publications. The mechanism file and thermochemical data for the current kinetic model are provided in the Supplementary Material.
All simulations in the present work are performed using the homogeneous reactor model of the COSILAB software. The nominal reaction time of 4.0 ms and constant pressure of 20 bar are used for the simulations. The assumption of constant pressure, typically used in simulations of speciation results from single-pulse shock tube experiments, has been well justified in previous publications (Han, Mehta, and Brezinsky Citation2019; Tang and Brezinsky Citation2006). However, it is suggested that certain reactions involving resonantly stabilized radicals may occur during the post-shock quenching period, resulting in an increase in the mole fractions of some products (Manion, Sheen, and Awan Citation2015; Mertens et al. Citation2018). In addition, depending on the characteristics of the experimental facility (small-bore shock tubes) or conditions (higher fuel concentrations) (Ferris, Davidson, and Hanson Citation2018; Mehta, Wang, and Brezinsky Citation2022), the constant-pressure assumption may be erroneous. Simulations with measured pressure profiles are performed over a longer time period (10.0 ms) for all n-heptane sets. The time-dependent concentrations of the major products and important radicals are shown in Figure S1 in the Supplementary Material. The plateaus in the concentration profiles of the main products are reached before the quenching process which indicates that their formation is already completed at the specified reaction time. The reaction systems currently studied do not produce large amounts of highly stabilized radicals such as propargyl and benzyl, so their concentrations are too low to significantly affect the amount of final products as shown in Figures S2-S4.
Similar to the vast literature on n-heptane experimental chemistry, several detailed kinetic models have been developed and optimized in the past decades based on the available data. Some of the most recent models, such as NUIG (Zhang et al. Citation2016), JetsurFv2 (Wang et al. Citation2010), CRECK (Pejpichestakul et al. Citation2019), and LLNL PAH (Shao et al. Citation2020), were also used to simulate the current experiments for comparison purpose (Figures S5–S7). The current model is compared to NUIG and JetsurFv2 for small hydrocarbon predictions, as well as the CRECK and LLNL PAH models for both small hydrocarbon and aromatic species predictions. In comparison to the other tested models, the LLNL PAH model is in good agreement with our experiments except for over-predictions for benzene, phenylacetylene, and naphthalene.
The current kinetic model for heptane pyrolysis is tested against pertinent speciation data reported in literature to assess its capability to predict results under different conditions (Garner, Sivaramakrishnan, and Brzezinski Citation2009; Held, Marchese, and Dryer Citation1997; Yasunaga et al. Citation2017, Citation2018; Yuan et al. Citation2011). There are a variety of experimental setups and, as a result, simulation methodologies involved. For the shock tube experiments (Garner, Sivaramakrishnan, and Brzezinski Citation2009; Yasunaga et al. Citation2017, Citation2018), each data point is simulated with the “homogeneous reactor” by entering the measured T5, p5 and reaction time. The “plug-flow” model is employed to simulate heptane pyrolysis speciation in a flow tube (Yuan et al. Citation2011). As for the experiments performed with the variable-pressure flow reactor (Held, Marchese, and Dryer Citation1997), an isobaric and adiabatic zero-dimensional reactor is used to simulate the species mole fraction time histories by providing the initial temperature, pressure, and chemical compositions. Figures S8–S12 in the Supplementary Material show the comparisons between the literature data and the simulations with the current model.
Results and discussions
In this section, experimental and simulated species concentrations as a function of post-shock temperature T5 will be compared to validate the prediction capabilities of the present kinetic model. Only the data from 502 and 2000 ppm of n-heptane are presented in the discussion, while the ones for 103 ppm are given in Figure S2 in the Supplementary Material. In general, the model can mimic the mole fraction profiles of observed pyrolysis species and capture their decomposition and formation temperature windows within the experimental errors. To gain insight into the chemistry of heptane degradation and aromatics growth, modeling analytical approaches such as rate of production (ROP) analyses and sensitivity analyses are applied.
Fuel decomposition and the formation of small hydrocarbons
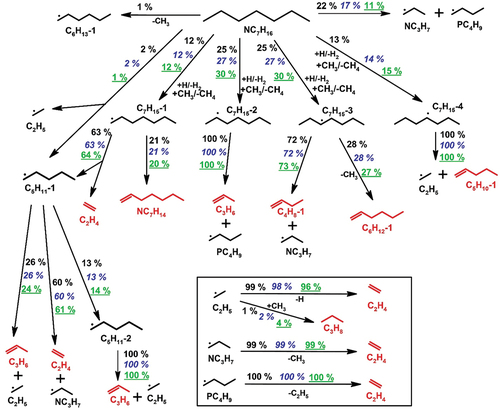
presents the experimental and simulated mole fraction profiles of n-heptane and C1–C6 acyclic species in the three separate sets. The kinetic model satisfactorily predicts the measured fuel conversion profiles as well as the formation of small intermediates in the individual studied cases. Olefins begin to form during the early stage of the fuel consumption. To determine the relationship between various alkene production and fuel consumption, displays initial fuel consumption pathways based on integrated rate-of-production (ROP) analyses over 4 ms in the three reaction systems. The temperatures chosen to perform these ROP calculations are 1160 K for 103 and 502 ppm heptane pyrolysis, and 1130 K for 2000 ppm heptane pyrolysis, where roughly half of the fuel is consumed in each case. The reaction scheme shows that n-heptane is largely consumed via the H-abstraction reactions, which produce 1-, 2-, 3-, and 4-heptyl radicals, and via the C-C bond scission reaction, which produces n-propyl (NC3H7) and 1-butyl (PC4H9) radicals. Other C-C bond fission reactions that produce methyl and 1-hexyl radicals, as well as ethyl and 1-pentyl radicals, contribute only slightly to the thermal decomposition of n-heptane. Following the decomposition of the major heptane radicals, the various alkene products are produced. Propylene (C3H6, ) and 1-pentene (C5H10-1, ) are directly formed through the unimolecular decomposition of 2- and 4-heptyl radicals, respectively (see ). The 3-heptyl radical allows a modest but significant fraction of methyl radical (CH3) and l-hexene (C6H12-1, ) to be formed in favor of NC3H7 and l-butene (C4H8-1, ). The allyl (C3H5-A)+CH3 recombination reaction is the second important C4H8-1 formation channel at 1160 K (around 30% according to the ROP analyses). C3H5-A is produced mostly from the unimolecular decomposition of C5H10-1 (C5H10-1= ethyl (C2H5) + C3H5-A) and C6H12-1 (C6H12-1= NC3H7+ C3H5-A), and partly from the C3H6 through H-abstraction reactions with methyl radical and hydrogen atom. The other butene isomer, 2-butene (C4H8-2, ), is produced mainly through isomerization and H-assisted isomerization of C4H8-1, with increasing contribution from C5H10-1 + CH3 reaction along with the initial fuel concentration (from 10% with 103 ppm to 30% with 2000 ppm initial n-heptane mole fraction at 1170 K). Ethylene (C2H4, ) is formed via numerous reaction pathways as shown in , including 1-heptyl decomposition, thermal decomposition of the 1-pentyl (C5H11-1), 1-butyl, l-propyl and ethyl radicals formed in 1-,2-,3-and 4-heptyl decomposition, respectively, and further H-abstraction reactions of the other olefinic intermediates, namely C3H6 and C4H8-1. The dominant C2H4 consumption route is the unimolecular fragmentation into vinyl radicals (C2H3) and H-atoms. Aside from alkenes, the recombination of methyl and ethyl drives propane formation (C3H8, ), which starts at the early phases of heptane decay. Pentadiene (LC5H8, ) is the result of the unimolecular decomposition of pentenyl (NC5H9-3) and heptenyl radicals (NC7H13), which are formed through dehydrogenation of C5H10-1 () and 1-heptene (NC7H14), respectively. NC7H14 (not measured in the current study) is produced entirely by the unimolecular breakdown of C7H15-1 ().
Figure 1. Measured (symbols) and simulated (lines) mole fraction profiles of fuel and small intermediates as a function of post-shock temperature T5 in heptane pyrolysis.
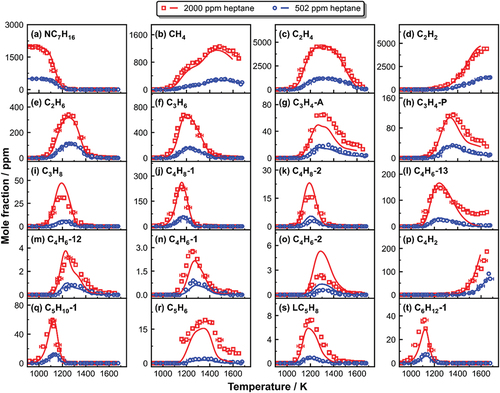
Figure 2. Fuel consumption pathways at 1160 K in 103 and 502 ppm n-heptane pyrolysis and at 1130 K in 2000 ppm n-heptane pyrolysis, 20 bar. The percentage numbers (103 ppm heptane: black normal; 502 ppm heptane: blue italic; 2000 ppm heptane: green underlined) are the contributions by the corresponding reactions to the consumption of the species on the source side. The reaction path analyses are based on the integrated ROP analyses over 4 ms.
The remaining small hydrocarbons are formed through subsequent reactions. The remarkable mole fractions of methane (CH4) and ethane (C2H6) in n-heptane pyrolysis ( b, e, respectively) arise from the abundant CH3 production via NC3H7 = CH3 + C2H4 reaction at low temperatures (>1200 K), H + C3H6 = CH3 + C2H4 reaction at moderate temperatures (1200–1350 K), and unimolecular decomposition of C2H6 at high temperatures (>1400 K). Acetylene (C2H2, ) is predominantly formed by the decomposition of C2H3 and marginally by the reaction of H atoms with allene (C3H4-A, ) and propyne (C3H4-P, ). The C3H4 isomers, C3H4-P and C3H4-A, are formed at temperatures where the fuel is nearly depleted. C3H6 consumption leads to C3H4-A, which isomerizes to C3H4-P. Four C4H6 isomers are observed: 1-butyne (1-C4H6, ), 2-butyne (2-C4H6, ), 1,2-butadiene (1,2-C4H6, ) and 1,3-butadiene (1,3-C4H6, ). Quantitative mole fraction profiles for C4H6 isomers are rarely reported in previous investigations on n-heptane pyrolysis. The current model captures 1,3-C4H6, 1,2-C4H6, and 1-C4H6 profiles well; nevertheless, discrepancies exist between the experimental and simulated 2-C4H6 speciation profiles with an overprediction of the experimental profiles. The peak mole fraction of 1,3-C4H6 is fifty times greater than that of the other three isomers. ROP analyses are done across the temperature formation windows to better explain the higher peak mole fractions of 1,3-butadiene. The formation of the dominant isomer 1,3-C4H6 largely relies on the 1-C4H8 consumption through the stepwise dehydrogenation via 1-buten-3-yl (C4H71–3) radical as an intermediate. As we observed above, 1-butene is one of the main alkenes from the direct decomposition of the heptyl radicals, thus its large concentrations () drive the formation of 1,3-C4H6. The other C4H6 isomers are mainly produced by bimolecular recombination reactions. 1,2-C4H6 is mainly formed through CH3 + propargyl (C3H3) reaction, where C3H3 is produced from the decomposition of C3H4-P and C3H4-A. Reactions between CH3 and C3 molecules/radicals (C3H4-A and C3H3) govern the formation of 1-C4H6. The production of 2-C4H6 mostly depends on the isomerization reactions of 1,2-C4H6 and just moderately by 1,3-C4H6 isomerization reactions. Regarding C5 species, cyclopentadiene (C5H6, ) is mainly produced through the dehydrogenation of cyclopentene (CYC5H8) and slightly through the reaction C3H5-A+C4H6=C2H5+C5H6. The 1-penten-4-yl radical (C5H914) formed by the C3H5-A+C2H4 recombination reaction can undergo cyclization reaction to produce cyclopentenyl radical (cyC5H9), which is the main source of CYC5H8 through its β-C – H scission. Though not shown in , CYC5H8 mole fractions are measured in our investigation and are available in the Supplementary Material.
Aromatic growth
depicts the experimental and simulated mole fraction profiles of several common mono-aromatic hydrocarbons (MAHs) and bicyclic and tricyclic PAHs. For all the aromatics, the current model accurately captures the experimental trends regarding the shapes and the peak concentrations. The major aromatic formation pathways are identified through ROP analyses and will be addressed in this section. The reactions that account for the formation of these aromatics are similar in all the reaction systems; however, the product concentrations increase with the fuel mole fraction due to the increased amount of carbon in the reaction system.
Figure 3. Measured (symbols) and simulated (lines) mole fraction profiles of MAHs and PAHs as a function of post-shock temperature T5 in n-heptane pyrolysis.
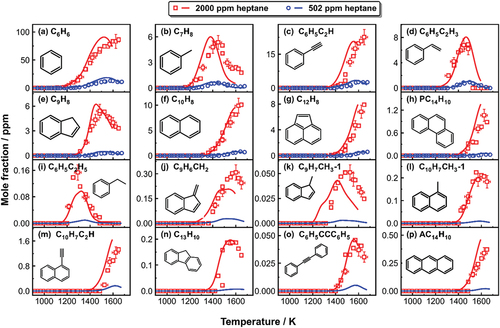
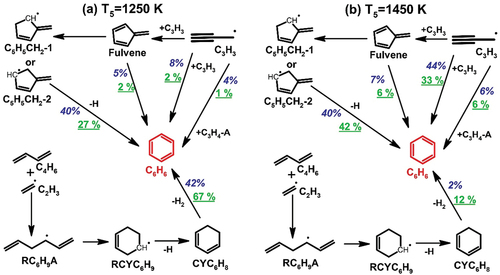
One of the key subjects covered in kinetic studies of acyclic fuel consumption under fuel-rich or pyrolytic conditions is the production of the first aromatic ring. The MAH species detected in n-heptane pyrolysis include benzene (C6H6), toluene (C7H8), phenylacetylene (C6H5C2H), styrene (C6H5C2H3), and ethylbenzene (C6H5C2H5). C6H6 () is the most abundant MAH species formed during n-heptane pyrolysis. Integrated ROP analyses at 1250 K and 1450 K are used to identify the reaction pathways leading to C6H6 production as shown in . Three sources are responsible for C6H6 production in heptane pyrolysis: the dehydrogenation of 1,4-cyclohexadiene (CYC6H8) that is mainly produced from C2H3+C4H6 recombination followed by cyclization, H-abstraction and β-scission (Wang, Villano, and Dean Citation2015), the reaction sequence 2C3H3 → fulvene→ C5H5CH2-1/C5H5CH2-2 →C6H6 (Miller and Klippenstein Citation2003), and the C3H3 self-recombination. The relative importance of the above-mentioned channels varies with temperature. The first two pathways control C6H6 formation in the low-temperature range of our study, whereas the latter two at higher temperatures. It is worth mentioning how the pathway through 1,4-cyclohexadiene is also strongly dependent on the initial fuel concentration. Besides, the C3H5-A+C3H3 recombination and the fulvene isomerization have minor contributions to C6H6 throughout the temperature range (see ).
Figure 4. The reaction pathways leading to benzene formation at (a) T5 = 1250 K, (b) T5 = 1450 K in the pyrolysis of n-heptane. The percentage numbers (502 ppm heptane: blueitalic; 2000 ppm heptane: green underlined) represent the contributions to benzene formation by the corresponding reactions.
The formation of C7H8 () largely depends on the reactions of C3H3 with C4H6 and but-2-yn-1-yl radical (C4H5-2), respectively. The formation of C6H5C2H () and C6H5C2H3 () mostly relies on the reactions of phenyl with C2H2/C2H and C2H4, respectively. C5H5+C3H3 reaction and C4H6 self-recombination also slightly contribute to C6H5C2H3 at low temperatures. C6H5C2H5 is mainly formed through benzyl (C7H7)+CH3. C4H6 self-recombination also has a non-negligible contribution to C6H5C2H5.
The major goal of this work is to unravel the mechanisms responsible for PAH formation during n-heptane pyrolysis. The smallest PAH detected in this study is indene (C9H8, ). C9H8 mainly originates from the bimolecular reaction C9H7+H=C9H8 at low temperatures (T5<1400 K), where C9H7 is exclusively formed from C4H2+C5H5 reaction. As the temperature increases, the isomerization of 1-phenyl-propyne (C6H5C3H3P_1) and phenyl-allene (C6H5C3H3A) and C7H7+C2H2 reaction become the dominant C9H8 formation channels. The corresponding rate parameters have been validated in our previous investigations on the pyrolysis of propylene and propyne (Sun et al. Citation2021a), and C2 and C3 addition to toluene and benzene (Hamadi et al. Citation2022; Sun et al. Citation2021a, Citation2022). C6H5C3H3P_1 is largely formed through the molecule+radical reaction C6H5C2H+CH3, whereas the radical+radical reaction C6H5+C3H3 is the major source of C6H5C3H3A.
C9H8 consumption mostly yields indenyl radical (C9H7) through H-abstraction and unimolecular decomposition. C9H7 participates in the formation of the other PAHs, including naphthalene (C10H8) and acenaphthylene (C12H8), which were found to be the most prevalent in the current study (). The formation mechanism of C10H8 includes several possible reaction pathways that are displayed in based on ROP analysis done at 1525 K. As can be seen in , benzofulvene (C9H6CH2) is a vital precursor of naphthalene. According to the ROP-analyzed results, the formation of C9H6CH2 relies on three major channels: i) the unimolecular decomposition of methyl-indene radical (C9H6CH3-1) following the production of 1-methyl-indene (C9H7CH3-1) through C9H7+CH3 recombination; ii) the recombination between fulvenallyl (C7H5) and C3H3. (C7H5 mainly comes from C7H7 decomposition and the C3H3+C4H2 recombination); iii) the bimolecular reaction between o-benzyne (o-C6H4) and vinylacetylene (C4H4). (o-C6H4 is mostly derived from C7H7 breakdown). Besides the isomerization of benzofulvene, other potential sources of naphthalene include the consumption of methyl-indene radicals (C9H7CH2 and C9H6CH3-1), C7H5+C3H3 reaction, and the hydrogen abstraction acetylene addition (HACA) route through phenylacetylene radical (C6H4C2H) leading to naphthyl (C10H7) radicals which will recombine with H atom (not shown in ).
Figure 5. The reaction pathways leading to naphthalene formation at T5 = 1525 K in the pyrolysis of heptane. The percentage numbers (502 ppm heptane: blue italic; 2000 ppm heptane: green underlined) represent the contributions to naphthalene formation by the corresponding reactions.
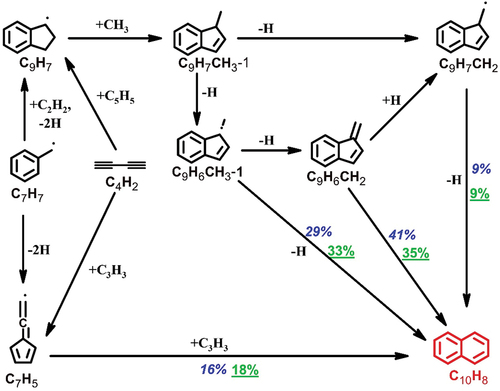
1-methyl-indene (C9H7CH3-1), 1-methyl naphthalene (C10H7CH3-1), acenaphthylene (C12H8), 1-ethynyl-naphthalene (C10H7C2H_1) and fluorene (C13H10) are among the major C10–C13 PAHs identified in this study. As previously stated, C9H7CH3-1 () is a key precursor for the naphthalene-benzofulvene isomer pair. The C9H7+CH3 recombination produces the majority of C9H7CH3-1. C12H8 () is one of the most abundant PAHs in n-heptane pyrolysis. The C9H7+C3H3 recombination and the subsequent dehydrogenation and ring-rearrangement processes (Jin et al. Citation2019) prevails C12H8 formation throughout the temperature range. The formation of the remaining PAHs involves the participation of naphthyl radical (C10H7-1): C10H7-1+CH3 recombination is the primary source of C10H7CH3-1 (); the HACA route C10H7-1+C2H2 = C10H7C2H_1+H controls the formation of C10H7C2H_1 () and it also partly accounts for C12H8 formation (27% at 1525 K); C10H7-1+C3H4-P is a minor route leading to C13H10 (). In this case, the C9H7+vinylacetylene (C4H4) reaction governs the C13H10 formation throughout the temperature range.
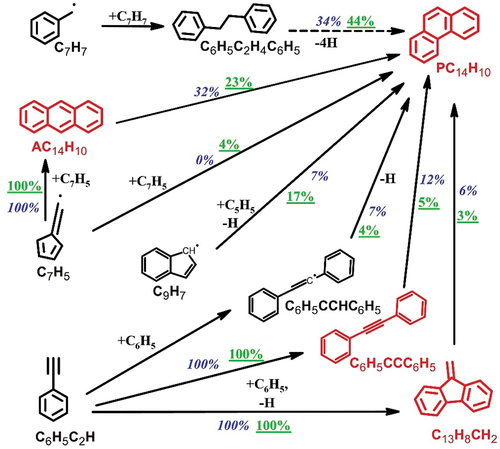
Four different C14H10 species are identified and quantified in 2000 ppm heptane pyrolysis, including the dominant phenanthrene (PC14H10), and its isomers, diphenylacetylene (C6H5CCC6H5), 9-methylene-fluorene (C13H8CH2), and anthracene (AC14H10). Reaction pathways leading to C14H10 isomers formation at 1500 K are presented in . C6H5CCC6H5 () and C13H8CH2 (not shown in ) are the products of the C6H5C2H+C6H5 addition-elimination reaction (Sun et al. Citation2020). AC14H10 () is totally formed from C7H5 self-recombination. PC14H10 () mainly comes from the C7H7 self-recombination and AC14H10 isomerization. Other reaction channels which contribute to PC14H10 formation include: the C7H5 self-recombination, H-assisted isomerization of C6H5CCC6H5 and C13H8CH2, and the reaction of C9H7 with cyclopentadienyl radical (C5H5), which results from the consumption of C5H6 and the addition of C3H3 to C2H2.
Figure 6. The reaction pathways leading to the formation of C14H10 isomers at T5 = 1500 K in the pyrolysis of n-heptane. The percentage numbers (502 ppm n-heptane: blue italic; 2000 ppm n-heptane: green underlined) represent the contributions to C14H10 isomers formation by the corresponding reactions. The dashed arrows represent multi-step reaction.
Conclusion
This research highlights PAH products from n-heptane pyrolysis as a significant new contribution to the literature on the chemistry of this important surrogate component. The experiments were carried out in a single-pulse shock tube coupled to gas chromatographic techniques at a nominal pressure of 20 bar throughout a temperature range of 900–1700 K. Updates to our ongoing PAH kinetic model demonstrate satisfactory predictive performances for the speciation measurements obtained in this work as well as those reported in literature studies on n-heptane pyrolysis. Heptane mainly decomposes through C-C fission and H-abstraction reactions leading to the formation of heptyl, n-butyl and n-propyl radicals. These Alkyl radicals further undergo beta-fission reactions leading to the formation of alkenes, namely, ethylene, propylene, 1-butene, 1-pentene and 1-hexene. The consumption of alkenes results in key intermediates such as methyl, acetylene, ethyl, propargyl, and 1,3-butadiene, which play a significant role in the formation and growth of aromatics. In particular, the formation of benzene relies on C2H3+C4H6 reaction through the intermediate cyclohexene, the H-addition and isomerization of fulvene, and C3+C3 reactions, more specifically, propargyl self-recombination and propargyl+allene reaction. Toluene is the product of the addition reaction of C3H3 to 1,3-butadiene. Indene formation mainly depends on C4H2+C5H5 reaction at low temperatures and on the consumption of C6H5C3H3P_1 and C6H5C3H3A, and the C7H7+C2H2 channel at high temperatures (>1400 K). Indenyl’s subsequent interactions with methyl, propargyl and vinylacetylene lead to the production of naphthalene, acenaphthylene and fluorene, respectively. Naphthyl radicals further participate in the formation of other bigger PAHs including 1-methyl naphthalene and 1-ethynyl naphthalene. Benzyl self-recombination and anthracene isomerization were analyzed to be the greatest contributors to phenanthrene formation. A small amount of phenanthrene is produced through C6H5C2H+C6H5 and C9H7+C5H5 reaction channels. Finally, the findings will help researchers better understand how PAHs form during the combustion of surrogate fuels.
SMM4_thermdata.dat
Download (385.4 KB)SMM3_mechanism.inp
Download (2.6 MB)SMM2_Experimental_data.xlsx
Download MS Excel (680.8 KB)SMM1_.docx
Download MS Word (2.6 MB)Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement n° 756785).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/00102202.2023.2182199
Additional information
Funding
References
- Appleby, W. G., W. H. Avery, and W. K. Meerbott. 1947. Kinetics and mechanism of the thermal decomposition of n-heptane. J. Am. Chem. Soc. 69 (10):2279–85. doi:10.1021/ja01202a012.
- Aribike, D., and A. Susu. 1988. Mechanistic modeling of the pyrolysis of n-heptane. Thermochim Acta 127:259–73. doi:10.1016/0040-6031(88)87502-6.
- Aribike, D. S., and A. A. Susu. 1988. Kinetics and mechanism of the thermal cracking of n-heptane. Thermochim Acta 127:247–58. doi:10.1016/0040-6031(88)87501-4.
- Bajus, M., V. Veselý, P. A. Leclercq, and J. A. Rijks. 1979. Steam cracking of hydrocarbons. 1. Pyrolysis of heptane. Resour. Policy 18 (1):30–37. doi:10.1021/i360069a007.
- Chakraborty, J. P., and D. Kunzru. 2009. High pressure pyrolysis of n-heptane. J Anal Appl Pyrolysis 1 (86):44–52. doi:10.1016/j.jaap.2009.04.001.
- Comandini, A., T. Malewicki, and K. Brezinsky. 2012. Online and offline experimental techniques for polycyclic aromatic hydrocarbons recovery and measurement. Rev Sci Instrum 83 (3):034101. doi:10.1063/1.3692748.
- Curran, H. J., P. Gaffuri, W. J. Pitz, and C. K. Westbrook. 1998. A comprehensive modeling study of n-heptane oxidation. Combust. Flame 114 (1):149–77. doi:10.1016/S0010-2180(97)00282-4.
- Davidson, D., M. Oehlschlaeger, and R. K. Hanson. 2007. Methyl concentration time-histories during iso-octane and n-heptane oxidation and pyrolysis. Proc Combust Inst 31:321–28. doi:10.1016/j.proci.2006.07.087.
- Ferris, A. M., D. F. Davidson, and R. K. Hanson. 2018. A combined laser absorption and gas chromatography sampling diagnostic for speciation in a shock tube. Combust. Flame 195:40–49. doi:10.1016/j.combustflame.2018.04.032.
- Garner, S., R. Sivaramakrishnan, and K. Brzezinski. 2009. The high-pressure pyrolysis of saturated and unsaturated C7 hydrocarbons. Proc Combust Inst 32:464–67. doi:10.1016/j.proci.2008.06.217.
- Hamadi, A., L. Carneiro Piton, S. Abid, N. Chaumeix, and A. Comandini. 2022. Combined high-pressure experimental and kinetic modeling study of cyclopentene pyrolysis and its reactions with acetylene. In Proc. Combust, Inst doi:10.1016/j.proci.2022.07.023.
- Hamadi, A., W. Sun, S. Abid, N. Chaumeix, and A. Comandini. 2022. An experimental and kinetic modeling study of benzene pyrolysis with C2−C3 unsaturated hydrocarbons. Combust. Flame 237:111858. doi:10.1016/j.combustflame.2021.111858.
- Han, X., J. M. Mehta, and K. Brezinsky. 2019. Temperature approximations in chemical kinetics studies using single pulse shock tubes. Combust. Flame 209:1–12. doi:10.1016/j.combustflame.2019.07.022.
- Held, T. J., A. J. Marchese, and F. L. Dryer. 1997. A semi-empirical reaction mechanism for n-heptane oxidation and pyrolysis. Combust. Sci. Technol. 123 (1–6):107–46. doi:10.1080/00102209708935624.
- Hugoniot, P. H. 1887. Sur la Propagation du Mouvement dans les Corps et Spécialement dans les Gaz Parfaits (première partie) 57:3–97.
- Hugoniot, P. H. 1889. Sur la Propagation du Mouvement dans les Corps et Spécialement dans les Gaz Parfaits (deuxième partie) 58:1–125.
- Jin, H., L. Xing, J. Hao, J. Yang, Y. Zhang, C. Cao, Y. Pan, and A. Farooq. 2019. A chemical kinetic modeling study of indene pyrolysis. Combust. Flame 206:1–20. doi:10.1016/j.combustflame.2019.04.040.
- Li, X., Z. Ma, E. Lv, Y. Dong, and X. Wang. 2021. Experimental and kinetic study of hydrocarbon fuel pyrolysis in a shock tube. Fuel 304:121521. doi:10.1016/j.fuel.2021.121521.
- Manion, J. A., D. A. Sheen, and I. A. Awan. 2015. Evaluated kinetics of the reactions of H and CH3 with n-alkanes: Experiments with n-butane and a combustion model reaction network analysis. J Phys Chem A 119 (28):7637–58. doi:10.1021/acs.jpca.5b01004.
- Mehta, J. M., W. Wang, and K. Brezinsky. 2022. Shock tube study of natural gas oxidation at propulsion relevant conditions. Proc Combust Inst. doi:10.1016/j.proci.2022.06.012.
- Mertens, L. A., I. A. Awan, D. A. Sheen, and J. A. Manion. 2018. Evaluated site-specific rate constants for reaction of isobutane with H and CH3: Shock tube experiments combined with bayesian Model optimization. J Phys Chem A 122 (49):9518–41. doi:10.1021/acs.jpca.8b08781.
- Miller, J. A., and S. J. Klippenstein. 2003. The recombination of propargyl radicals and other reactions on a C6H6 potential. J Phys Chem A 107 (39):7783–99. doi:10.1021/jp030375h.
- Murata, M., and S. Saito. 1974. Prediction of initial product distribution from n-paraffin pyrolysis at higher temperatures by considering ethyl radical decomposition. J. Chem. Eng. Jpn 7 (5):389–91. doi:10.1252/jcej.7.389.
- Murata, M., and S. Saito. 1975. A simulation model for high-conversion pyrolysis of normal paraffinic hydrocarbons. J. Chem. Eng. Jpn 8 (1):39–45. doi:10.1252/jcej.8.39.
- Pant, K. K., and D. Kunzru. 1996. Pyrolysis of n-heptane: Kinetics and modeling. J Anal Appl Pyrolysis 36 (2):103–20. doi:10.1016/0165-2370(95)00925-6.
- Pejpichestakul, W., E. Ranzi, M. Pelucchi, A. Frassoldati, A. Cuoci, A. Parente, and T. Faravelli. 2019. Examination of a soot model in premixed laminar flames at fuel-rich conditions. Proc Combust Inst 37 (1):1013–21. doi:10.1016/j.proci.2018.06.104.
- Pilla, G., D. Davidson, and R. Hanson. 2011. Shock tube/laser absorption measurements of ethylene time-histories during ethylene and n-heptane pyrolysis. Proc Combust Inst 33:333–40. doi:10.1016/j.proci.2010.06.146.
- Pyun, S., W. Ren, D. Davidson, and R. Hanson. 2013. Methane and ethylene time-history measurements in n-butane and n-heptane pyrolysis behind reflected shock waves. Fuel 108:557–64. doi:10.1016/j.fuel.2012.12.034.
- Rankine, W. J. M. 1870. XV. On the thermodynamic theory of waves of finite longitudinal disturbance. Philos. Trans. R. Soc. Lond 160:277–88.
- Shao, C., G. Kukkadapu, S. W. Wagnon, W. J. Pitz, and S. M. Sarathy. 2020. PAH formation from jet stirred reactor pyrolysis of gasoline surrogates. Combust. Flame 219:312–26. doi:10.1016/j.combustflame.2020.06.001.
- Sun, W., A. Hamadi, S. Abid, N. Chaumeix, and A. Comandini. 2020. An experimental and kinetic modeling study of phenylacetylene decomposition and the reactions with acetylene/ethylene under shock tube pyrolysis conditions. Combust. Flame 220:257–71. doi:10.1016/j.combustflame.2020.06.044.
- Sun, W., A. Hamadi, S. Abid, N. Chaumeix, and A. Comandini. 2021a. A comprehensive kinetic study on the speciation from propylene and propyne pyrolysis in a single-pulse shock tube. Combust. Flame 231:111485. doi:10.1016/j.combustflame.2021.111485.
- Sun, W., A. Hamadi, S. Abid, N. Chaumeix, and A. Comandini. 2021b. Detailed experimental and kinetic modeling study of toluene/C2 pyrolysis in a single-pulse shock tube. Combust. Flame 226:129–42. doi:10.1016/j.combustflame.2020.11.044.
- Sun, W., A. Hamadi, S. Abid, N. Chaumeix, and A. Comandini. 2021c. Probing PAH formation chemical kinetics from benzene and toluene pyrolysis in a single-pulse shock tube. Proc Combust Inst 38 (1):891–900. doi:10.1016/j.proci.2020.06.077.
- Sun, W., A. Hamadi, S. Abid, N. Chaumeix, and A. Comandini. 2022. Influences of propylene/propyne addition on toluene pyrolysis in a single-pulse shock tube. Combust 236:111799. doi:10.1016/j.combustflame.2021.111799.
- Sun, W., A. Hamadi, F. E. C. Ardila, S. Abid, N. Chaumeix, and A. Comandini. 2022. Insights into pyrolysis kinetics of xylene isomers behind reflected shock waves. Combust. Flame 244:112247. doi:10.1016/j.combustflame.2022.112247.
- Tang, W., and K. Brezinsky. 2006. Chemical kinetic simulations behind reflected shock waves. Int. J. Chem. Kinet. 38 (2):75–97. doi:10.1002/kin.20134.
- Wang, H., E. Dames, B. Sirjean, D. A. Sheen, R. Tango, A. Violi, J. Y. W. Lai, F. N. Egolfopoulos, D. F. Davidson, R. K. Hanson, et al. 2010. High-temperature chemical kinetic model of n-alkane (up to n-dodecane), cyclohexane, and methyl-, ethyl-, n-propyl and n-butyl-cyclohexane oxidation at high temperatures 2(2): 19. JetSurF version 2.0.
- Wang, K., S. M. Villano, and A. M. Dean. 2015. Reactions of allylic radicals that impact molecular weight growth kinetics. Phys. Chem. Chem. Phy 17 (9):6255–73. doi:10.1039/C4CP05308G.
- Yasunaga, K., S. Etoh, H. Yamada, H. Oshita, and Y. Hidaka. 2018. Modeling and experimental study on pyrolysis of isooctane and n-heptane behind reflected shock waves. Chem. Lett. 47 (6):747–50. doi:10.1246/cl.180154.
- Yasunaga, K., H. Yamada, H. Oshita, K. Hattori, Y. Hidaka, and H. Curran. 2017. Pyrolysis of n-pentane, n-hexane and n-heptane in a single pulse shock tube. Combust. Flame 185:335–45. doi:10.1016/j.combustflame.2017.07.027.
- Yuan, T., L. Zhang, Z. Zhou, M. Xie, L. Ye, and F. Qi. 2011. Pyrolysis of n-heptane: Experimental and theoretical study. J.Phys. Chem. A 115 (9):1593–601. doi:10.1021/jp109640z.
- Zamostny, P., Z. Bělohlav, L. Starkbaumová, and J. Patera. 2010. Experimental study of hydrocarbon structure effects on the composition of its pyrolysis products. J Anal Appl Pyrolysis 87 (2):207–16. doi:10.1016/j.jaap.2009.12.006.
- Zhang, K., C. Banyon, J. J. Bugler, H. Curran, A. Rodriguez, O. Herbinet, F. Battin Leclerc, C. B’chir, and A. Karl. 2016. An updated experimental and kinetic modeling study of n-heptane oxidation. Combust. Flame 172:116–35. doi:10.1016/j.combustflame.2016.06.028.