
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The gamma ray radionuclides Cs-137, Ba-140, I-131, Ce-141, Ru-103, Zr-95, and Np-239 were produced by neutron irradiation of UO2–ZrO2 solid solutions that were synthesized as simulated fuel debris under reducing and oxidizing conditions. The leaching ratio of radionuclides was investigated under atmospheric conditions at 25 °C for non-filtered natural surface seawater, as well as deionized water after filtration with a membrane of 0.45-µm pore size or that of nominal molecular weight limit of 3 kDa. The uranium molar concentration was affected by the oxidation state in the solid solution samples. The congruent dissolution of Cs, I, and Ba with the hexavalent uranium of U3O8 was facilitated in the seawater samples, whereas a lower leaching ratio of nuclides was observed in the deionized water samples. Neptunium-239, originally produced from uranium-238 in U3O8, showed behavior that was similar to that of Cs, I, and Ba. However, the dissolution of Np (as a parent nuclide of Pu-239) in the debris of UO2 and UO2–ZrO2 was suppressed in the same manner as Zr(IV) and Ce(IV). The concentration exhibited no filtration dependence after 15 d, which shows that most of the leached nuclides can exist in their ionic form in seawater.
1. Introduction
After the station blackout of the Tokyo Electric Power Company (TEPCO) Fukushima Daiichi Nuclear Power Station (NPS), thousands of tons of seawater were injected to cool the high-temperature reactor cores. Under the high-temperature conditions in the pressure vessel, uranium and several kinds of radioactive material – in the form of fission products (FPs) and actinide elements – are thought to have reacted with a certain amount of the zircaloy (Zry) in the fuel cladding and/or its oxide, ZrO2. Extensive experimental investigations have been performed to understand the high-temperature chemical interaction between Zry and fuel, and to study the severe accident (SA) analysis codes [Citation1]. Since the SA in Fukushima, it has become necessary to obtain knowledge of the distribution of radioactive material inside the reactors and the status inside the damaged cores in order to proceed safely and efficiently with the process of decommissioning. Recently, Tanaka et al. experimentally evaluated the fractional release rate of FPs into the gas phase from the fuel with molten Zry, and discussed this in relation to the characteristics of liquefied fuel [Citation2].
Meanwhile, it must be noted that the cooling water injected into the reactor cores in Fukushima NPS was highly contaminated by the radioactive materials [Citation3]. In order to reduce the high gamma radioactivity of Cs in the contaminated water (which contained sea salt), the zeolite column system KURION was installed as an ion exchanger [Citation4,Citation5]. A similar treatment system was also utilized to clean up highly radioactive wastewater at the Three Mile Island Unit II reactor in Middletown, PA [Citation6], to separate Cs from high-level radioactive defense waste in Hanford, WA [Citation7], and to aid in nuclear waste processing [Citation8]. The zeolite sorbent has proven to be a great absorbent material for cesium. A co-precipitation system introduced by Areva decontamination instruments was operated mainly to decontaminate Cs and Sr by forming nickel ferrocyanide and barium chloride (to be a mixed crystal with sulfate) precipitations, respectively [Citation9]. The secondary wastes, which contained several kinds of radionuclides, were discharged to the temporary storing facility at Fukushima NPS. Although the present systems in Fukushima NPS could adsorb other heavy metal ions, trace amounts of minor FPs and actinides might still be retained in the waste. Prior to predisposal treatment, detailed information about the waste – i.e., the composition of nuclides, their chemical conditions, their quantity, etc. – must be obtained. Nevertheless, the radionuclide concentration has not been estimated yet because of the presently high radioactivity, among other issues.
Corrosion due to spent UO2 fuel and accompanying radionuclide releases into aqueous solution have been reported in earlier studies. The release rate from spent fuels to deionized water and artificial aqueous phases – such as NaCl solution – depended on the chemical components of solutions [Citation10]. The impact of the chemical conditions – such as pH, redox potential, saline concentration, and carbonate concentration – on the release rate is a critical issue that must be considered in comprehensive long-term safety assessments in countries planning spent fuel disposal [Citation11–13]. Such knowledge is useful for evaluating the radionuclide release from the melted core in Fukushima NPS into saline water. However, non-filtered surface seawater, which contains a variety of matrix ions, suspended matter, and impurities, is importantly different from the ideal and clear artificial solution used in labs. Thus, simulating radionuclide behavior in non-filtered seawater samples will be helpful in the forthcoming data analysis of minor FPs and actinides in contaminated water, and in managing the associated secondary wastes.
In our recent study [Citation14], possible fuel debris in the reactor under aerobic SA conditions – mainly composed of uranium and zirconium oxides – was synthesized; thermal neutrons were irradiated onto the simulated debris sample to produce FPs and actinides. After long-term static leaching (i.e., 160 d) in non-filtered seawater under aerobic conditions, the gamma ray fission products Cs-137, Ru-103, Zr-95, and uranium in UO2–ZrO2 solid solutions were observed in the leachate. However, short-lived nuclides have already decayed out at this point. In the present work, we focus on the leaching behavior of shorter half-life fission products in order to investigate a wider variety of radionuclides. Furthermore, we discuss the leaching fractions of nuclides in relation to that of uranium. A leaching test in a Milli-Q water system is adopted as a reference for comparison with the seawater case. The formation of polymeric and colloidal species is also studied in order to quantify the membrane filter pore size dependence (3 kDa and 0.45 μm) on the filtrate concentration.
2. Experimental
2.1. Preparation of UO2–ZrO2 solid solution samples
According to the pseudo-binary diagram of the UO2–ZrO2 system [Citation15], three main regions – (Equation1(1)
(1) ) face-centered cubic (fcc), (2) fcc + tetragonal, and (3) tetragonal at 1473 K – exist with the increasing ZrO2 fraction. The borders between (Equation1
(1)
(1) ) and (2) and between (2) and (3) appear at ∼11 and ∼84 mol% ZrO2, respectively. Therefore, three different compositions of UO2–ZrO2 samples were adopted to be 10, 50, and 90 mol% ZrO2. Uranium dioxide powder was obtained by reducing U3O8 powder using Ar with 10% H2 gas at 1473 K for 4 h. UO2 samples with 0 mol% ZrO2 were also prepared for comparison. First, the weighed amounts of UO2 and ZrO2 powders were mixed in a zirconia pod with 14 zirconia balls in an argon-filled glove bag. The pot was then covered with a zirconia cap and sealed with aluminum tape. The UO2–ZrO2 mixture was obtained by the mechanochemical method using a Fritsch Pulverisette planetary mill with a rotation speed of 500 rpm. After the mechanochemical treatment, the solid solution samples were prepared by heating at 1473 K for 1 h in a reducing (Ar + 10% H2) or oxidizing (N2 + 2% O2) atmosphere.
X-ray diffraction (XRD) measurements of the products were conducted using a Rigaku Ultima-IV system with Cu–Kα irradiation (40 kV, 40 mA). Though the surface area measurement of the neutron-irradiated solid solution samples is helpful in evaluating the absolute leaching ratio of each element from the solid solution samples, it could not be conducted owing to laboratory regulations. However, the surface area data are not needed to discuss the leaching behavior in the present study because the leaching ratio of nuclides is normalized to that of the matrix of uranium in each sample. In fact, Kirishima et al. observed the morphology of a solid solution after 22 h of heat treatment at 1373 K in an Ar atmosphere using a scanning electron microscope (SEM) [Citation16]. The SEM image indicated that small unit particles having diameter less than or equal to 1 μm coagulate with each other, and form ∼10 μm grains.
2.2. Static leaching test of radionuclides in solid solutions to seawater
About 30 mg of the UO2–ZrO2 solid solution sample was sealed in a quartz tube in vacuo. The tube was inserted into a polyethylene capsule and irradiated using a pneumatic transferring system (Pn-2) of the Kyoto University Reactor at a thermal neutron flux of 5.5 × 1012 neutrons cm−2 s−1 (at 1 MW) for 20 min. The gamma ray activity as an initial inventory of each radionuclide in the solid sample was measured by using a Ge detector (GMX15P4, Ortec) after one week of cooling in order to allow for the decay of highly radioactive, short-lived nuclides.
The irradiated sample and 50 mL of the non-filtrated surface seawater (or ultrapure Milli-Q deionized water) were placed in a 50-mL polypropylene bottle to perform leaching tests. The seawater was collected using a hand pump from the seashore at the Urajiri coast in Odaka-ku, Minamisoma, Fukushima, 11 km north from the Fukushima Daiichi NPS. It was thereafter stored in a cool, dark place. Most of the visible suspended matter, such as coarse sand, settled out from gravity. The composition of the major elements of the seawater used in the tests was measured and compared with reference data of surface water from the North Pacific Ocean in our previous study [Citation16], so that the seawater used in this study can be regarded as typical surface seawater having an ionic strength of ∼0.7 [Citation17]. The concentration of natural uranium (as UO2(CO3)34−) was reported as low as 1.35 × 10−8 mol dm−3 (M) [Citation18]. This value is lower than those in the present study as discussed later.
During the static leaching test under atmospheric conditions at 25 °C, the bottles were shaken vigorously by hand once per week. After being subjected to leaching, 5 mL of the supernatant was filtrated by a membrane with a 0.45-µm pore size (Advantec) or a membrane with a nominal molecular weight limit of 3 kDa (Microcon, Millipore), followed by transport to a new polypropylene test tube. The filtrate was evaporated at 363 K in order to prepare a solidified point-like source of gamma rays. The spectra were acquired for around 24 h, and the photo peak area corresponding to each nuclide was evaluated, taking into account a background reduction. The uranium concentration was determined by using inductively coupled plasma mass spectrometry (ICP-MS, Element2, Thermo Sci.). The cumulative radioactivity of the nuclides leached during the 28-d period was determined by considering a decay correction, according to the previous study [Citation14]. The leaching ratio RM of element ‘M’ is defined as
(1)
(1) where Ai expresses the initial inventory (Bq) initially present in the solid solution sample, Af expresses the radioactivity (Bq) in the filtrate measured after the leaching period (t), and t1/2 expresses the half-life of the nuclide. The molar concentration of uranium was used to calculate the RU value.
3. Results and discussion
3.1. Solid solutions
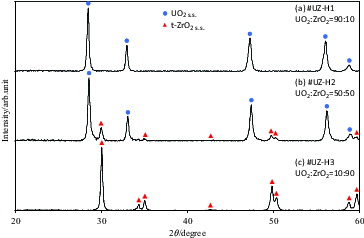
After the mechanochemical treatment of UO2–ZrO2 mixtures with different UO2/ZrO2 ratios, the peaks for the UO2 phase shifted to higher angles with an increasing Zr ratio, whereas those for the monoclinic ZrO2 appeared at the 90 mol% ZrO2 sample. After this stage, the samples were heat treated in both reducing and oxidizing atmospheres. shows the XRD patterns of samples with different UO2/ZrO2 ratios after heat treatment at 1473 K in Ar + 10% H2 for 1 h. In the case of the 90:10 sample ( (a)), only the XRD pattern with sharpened peaks, which correspond to the UO2 fluorite structure, was observed. The lattice parameter of this phase was smaller than that of UO2. In , peaks for the fluorite structure slightly shifted to higher angles. The lattice parameter of this fluorite phase linearly decreased with an increasing zirconium ratio, showing the formation of a solid solution. In this figure, small peaks corresponding to the t-ZrO2 phase, which is the high-temperature structure of ZrO2, are also observed. The peaks for this t-ZrO2 phase become dominant, whereas those for the fluorite phase seem to disappear in .
Figure 1. X-ray diffraction (XRD) patterns of UO2–ZrO2 samples with different UO2/ZrO2 ratios after heat treatment at 1473 K in Ar + 10% H2 for 1 h. Circles represent UO2 solid solution (s.s.), and triangles represent t-ZrO2.
The XRD patterns of samples with different UO2/ZrO2 ratios after heat treatment at 1473 K in N2 + 2% O2 for 1 h are shown in . It can be seen that the U3O8 phase is dominant for the uranium-rich samples, and the Zr(IV) did not make a solid solution. With an increasing zirconium ratio, however, the m-ZrO2 phase appeared. Based on the XRD analysis, the predominant phase conditions are summarized in . When the UO2–ZrO2 (50:50 mol%) sample was heat treated under reducing conditions, two types of solid solutions (s.s.) co-existed – i.e., the fcc UO2 s.s. with zirconium (ZryU1−yO2−x), and the tetragonal ZrO2 s.s. with uranium (t-UyZr1−yO2−x). At a higher fraction of Zr (#UZ-H3), the t-ZrO2 s.s. was dominant, and the UO2 s.s. phase was observed at a higher fraction of U (#UZ-H1). Meanwhile, under oxidative conditions with 2% O2, the higher oxidation phase, U3O8, was observed with t-ZrO2 (#UZ-O1), but UO2 s.s. still remained in the UO2–ZrO2 sample (#UZ-O2), suggesting that tetravalent UO2 might be partially stabilized in the solid solution with ZrO2.
Figure 2. X-ray diffraction (XRD) patterns of UO2–ZrO2 samples with different UO2/ZrO2 ratios after heat treatment at 1473 K in N2+2% O2 for 1 h. Diamonds represent U3O8, squares represent t-ZrO2, and triangles represent m-ZrO2.
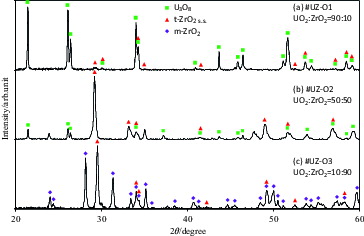
Table 1. Properties of solid solution samples: major phase conditions, initial inventory of nuclides Ai, and the half-life and the energy of peaks assigned.
3.2. Inventory of radionuclides in solid solutions
For the inventory evaluation of the radionuclides, the gamma ray spectrum of neutron-irradiated solid solution samples was measured prior to the leaching test. The peaks for seven radionuclides – Zr-95, Ru-103, I-131, Cs-137, Ba-140, Ce-141, and Np-239 – were assigned. Ru, I, Cs, Ba, and Ce were produced by the fission reaction of uranium-235, and neptunium-239 was converted by the neutron capture of the fertile isotope uranium-238 followed by beta-decay. Zr-95 was produced from both the fission reaction of uranium and the (n, γ) reaction of Zr-94 in the ZrO2 matrix, suggesting that Zr-95 from two different origins would exist in the #UZ-H and #UZ-O samples. Much shorter half-life nuclides – such as I, Ba, and Np – are discussed in the present study, whereas only Zr-95, Ru-103, and Cs-137 were discussed in the previous study [Citation14]. La-140 (t1/2 = 1.68 d) in radioactive equilibrium of Ba-140 was not considered. The initial inventory Ai and the half-life of gamma-emitting radionuclides are shown in .
3.3. Time dependence of radionuclide concentration leached
The pH and Eh values of the seawater and Milli-Q water under aerobic conditions were almost constant for every sample. More specifically, the values pH = 7.71 ± 0.18 and 6.20 ± 0.14, as well as Eh = +0.434 ± 0.068 and +0.507 ± 0.013 (V vs. NHE), were obtained for seawater and Milli-Q water, respectively.
3.3.1. Uranium
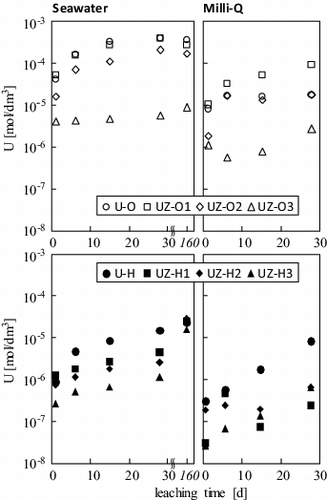
The matrix uranium molar concentration in the filtrates is shown in . The concentration for #U-O (100:0 of U/Zr mol%) in natural seawater was the highest at over 10−4 M, which is close to the concentration after 160 d of leaching [Citation14]. A similar behavior was observed for #UZ-O1 and #UZ-O2 (90:10 and 50:50 of U/Zr, respectively). This suggests that the uranyl ion was dissolved from the oxidized uranium compound, U3O8, as assigned by XRD spectrometry (). Such a high molar concentration of uranium could be explained by considering the carbonate and hydroxo complexes – e.g., UO2(OH)(CO3)− and (UO2)3(OH)5+ – and the solubility-limiting solid phase of amorphous UO3·2H2O at neutral pH and atmospheric conditions in seawater [Citation14]. However, the uranium leached from #UZ-O3 (10:90 of U/Zr) was largely suppressed into the 10−6 M range, while the concentration is two orders higher than the reference concentration in natural seawater. This suppression is most likely due to the tetravalent UO2 s.s. as the predominant phase (). On the other hand, the #U-H and #UZ-H series show low uranium concentrations, and the uranium concentration up to 28 d strongly depends on the UO2/ZrO2 ratios. The leaching of the #UZ-H series would not achieve equilibrium, and the concentrations reach 10−5 M after 160 d. Hence the uranium concentration leached from UO2 s.s. of the #UZ-H series might be controlled by uranium oxidation reactions at the surface of sample powders. As shown in , the lower uranium concentration in Milli-Q water also derives from a different oxidation process of UO2 s.s., suggesting that the tetravalent uranium was oxidized not only by dissolved oxygen, but also by other minerals, such as oxidants and microorganisms in the seawater.
Figure 3. Time dependence of the uranium molar concentration after filtration through a 0.45-μm-pore-size membrane in seawater (left) and Milli-Q water (right).
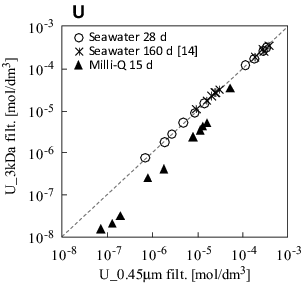
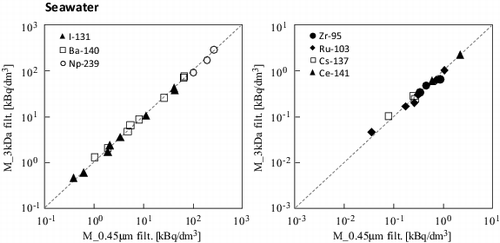
There may be another reason for the lower molar concentration of uranium. shows the filtration effect on the concentration through 3 kDa and 0.45-μm membranes. For the seawater samples, at 28 and 160 d, no significant dependence of the membrane pore size on the uranium concentration was observed, regardless of the type of solid solution under question. This is explained by the fact that uranium carbonate complex ions are predominant [Citation14]. However, the concentration after 3 kDa filtration of Milli-Q water decreased by 58% ± 4%, suggesting a promotion of nano-sized uranium colloid formations in the Milli-Q water. The uranium ions at a lower carbonate concentration and a neutral pH might form the colloidal hydrolysis species as eigencolloids.
Figure 4. Uranium molar concentrations after filtration through 3 kDa and 0.45-μm-pore-size membranes.
3.3.2. Fission products
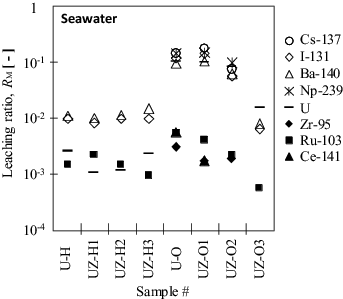
Multiple peaks of six FPs were observed in the gamma ray spectra of filtrates that passed through the 0.45-μm membrane (). The ratio after 15 d of leaching into seawater, RM, is shown in . Some plots are lacking because of the detection limit of gamma ray spectrometry. In the seawater samples, the RM values of the alkali metal ion Cs-137, the halide ion I-131, and the alkaline earth metal ion Ba-140 for #U-O and #UZ-O1/-O2 are high at around 10−1. It should be noted that, on the other hand, most of the RM values in the Milli-Q system were not determined owing to the significantly low radioactivity of filtrates. The uranium as the solid matrix was oxidized in preparation for the neutron irradiation. The values are one order higher than those for the #U-H and #UZ-H series containing UO2 s.s., which is more insoluble in an aqueous phase than U3O8. The low RM values for tetravalent zirconium and cerium in the #U-O and #UZ-O series after the filtration with the 0.45-μm membrane show the same tendency as ruthenium. The molar concentrations calculated from RZr and RCe are very low, and it would be unlikely that a water-insoluble metal complex would form and precipitate on the surface of the melted core. Such leaching behavior is discussed later with respect to the relationship between RM and RU.
Figure 5. Typical gamma ray spectra measured for the filtrate of #U-O (15 d of leaching in seawater), which was passed through the 0.45-μm membrane. The characteristic gamma ray emitters for quantification are marked above the corresponding peaks with underlining.
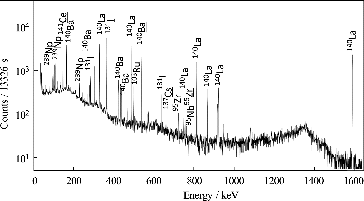
Figure 6. Leaching ratio RM of the radionuclides and uranium leached in seawater after 15 d.
shows the time dependence of the ratio RM of FPs normalized by that of the corresponding uranium, RU, in the seawater samples. As described above, RM/RU values for the Milli-Q water could not be determined owing to low radioactivities.
Figure 7. Time dependence of RM/RU in the seawater sample system; #U-O and #UZ-O series (upper) and #U-H and #UZ-H series (lower).
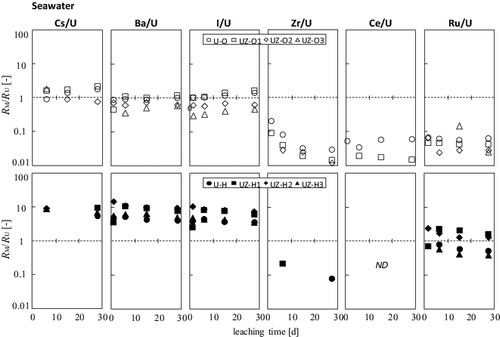
In the #U-O and #UZ-O series, the RM/RU values of Cs, Ba, and I remain constant at about 1. This fact can be explained by the congruent dissolution with hexavalent uranium of U3O8. Meanwhile, the low RM/RU values of tetravalent Ru and Ce ions remain in the range of 0.1–0.01, resulting from the low RM, while the hexavalent uranium was leached. A similar tendency was observed for Zr. As shown in , the atomic abundance of the Zr-95 produced via fission to the neutron-activated Zr-95 depends on the fraction in solid solutions. For #U-O, #UZ-O1/-O2 with higher Zr abundance (), most of the Zr produced via fission would be produced in U3O8, whereas that in the structure of UO2 s.s. should be a minority; that is to say, activated zirconium atoms in the ZrO2 structure are predominant for #UZ-O3. Since the RZr/RU values are similar to each other, the present results imply that the two origins of Zr-95 can hardly be distinguished from the chemical–structural point of view. As suggested in the previous work [Citation14], the concentration of Zr-95 leached was much higher than the estimated solubility of ZrO2(cr), but it is roughly comparable to the solubility limit of Zr(OH)4 (i.e., ∼10−14 M), indicating the possible formation of amorphous Zr hydroxide at the sample surface [Citation19].
In the #U-H and #UZ-H series, the RM/RU values of Cs, Ba, and I were constant with time, but were one order higher than those for the #U-O and #UZ-O series. These trace (but highly soluble) nuclides would be preferentially leached from the bulk surface of UO2 s.s. under a mild oxidation process of uranium. Most of the RZr and RCe values could not be determined because of the fact that they fell below the detection limit. However, since the leaching of Ru from the #U-H and #UZ-H series was congruent with that of tetravalent uranium oxide, the oxidation state of Ru in UO2 and UO2–ZrO2 may be different from that in U3O8.
No systematic explanation can be provided for the dependency of the different initial UO2/ZrO2 ratios on these RM/RU ratios. The analytical information of the oxidation states and the chemical structure at the powder surface of solid solutions is required for further consideration.
3.3.3. Neptunium
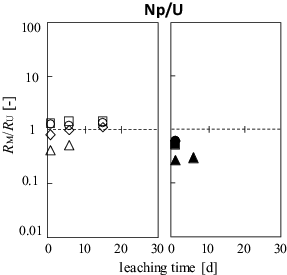
Minor actinide Np-239 shows a different leaching behavior from the FPs. The RNp/RU value in the #U-O and #UZ-O series remains close to 1 for a period of up to 15 d (); data points beyond 28 d are lacking because of the short half-life (t1/2 = 2.356 d). This leaching behavior is similar to that of water-soluble Cs, Ba, and I ions (). For the #U-H and #UZ-H series, however, the RNp/RU value is relatively low. This clearly differs from that of Cs, Ba, and I, but is similar to that of tetravalent Zr and Ce. The distinct leaching behavior of neptunium-239 can be explained by the oxidation state of neptunium at the sample surface. In the preparation of samples under reducing and oxidizing conditions, the predominant oxidation state of Np might be tetravalent and pentavalent in the UO2 s.s. and U3O8 matrices, respectively. The UO2 s.s. was mildly oxidized to UO22+, followed by a dissolution as discussed above. Though the Np(IV) is also expected to be oxidized to soluble Np(V), it should be noted that the standard oxidation–reduction potential of Np(IV/V) is more negative compared with that of U(IV/VI) [Citation20]. The different leaching behaviors of Np and U for the #U-H and #UZ-H series are most likely due to such a difference in potential.
Figure 8. Time dependence of the leaching ratio RNp normalized by that of RU in the seawater samples.
At the facilities of Fukushima Daiichi NPS, trace amounts of plutonium have been detected in the stagnant contaminated water samples collected [Citation21]. This suggests that plutonium should be contained in the supernatant of cooling water, and should be chemically captured by external water-cooling systems such as KURION and SARRY [Citation22]. Therefore, the leaching test of plutonium in the simulated solid solution samples promises to provide direct information of plutonium leaching behavior, and the trace Pu was detected in the leaching samples [Citation21]. Meanwhile, it should be recalled that Pu-239 can be produced by the beta decay of the pentavalent Np-239 leached into the aqueous solution. Corresponding to the RNp values of 1.47×10−1 and 6.85×10−2 for #UZ-O1 (15 d) and #UZ-H3 (6 d), respectively, the molar concentrations of Np were calculated to be 9.05×10−14 and 1.04×10−16 M, respectively. Supposing that all the Np-239 decayed with the short half-life, the radioactivity concentrations of Pu-239 would be 4.96×10−2 and 5.71×10−5 Bq dm−3, respectively. It is worth mentioning that the higher concentration for the oxidized sample is comparable to the combined values of Pu-239 and Pu-240 (e.g., (4.6 ± 0.8) × 10−1 Bq/dm3 [Citation21]) in the stagnant, contaminated water of Fukushima Daiichi NPS.
3.4. Dependence of filter membrane pore size on apparent solubility
shows the radioactivity of all seven nuclides after filtration through 3 kDa and 0.45-μm-pore-size membranes in the seawater samples. The dependence of the membrane pore size on the radioactive concentration was not observed regardless of the variety of solid solution under consideration as same as the dependence observed for uranium in , suggesting that no nano-sized colloidal species existed in the seawater after leaching for 15 d. However, the concentration for Ru-103 after 3 kDa filtration decreased by 18% ± 10% after the long period of 160 d [Citation14]. Though this fact implies a colloidal formation of Ru at a very low molar concentration in the seawater, the authors did not observe a similar behavior in the other short-lived nuclides, such as Zr-95, after 160 d [Citation14]. The colloid formation mechanism should, therefore, be elucidated in further experimental studies.
Figure 9. Radioactivity concentration after filtration through 3 kDa and 0.45-μm-pore-size membranes in the seawater samples after 15 d.
4. Conclusion
The leaching behaviors of trace amounts of Zr-95, Ru-103, I-131, Cs-137, Ba-140, Ce-141, and Np-239 produced by the neutron irradiation of simulated UO2–ZrO2 solid solutions in non-filtered seawater and Milli-Q water were related to the initial oxidation state of uranium in the samples. The hexavalent uranium ions in U3O8 dissolved easily, whereas the oxidation reaction of tetravalent uranium in UO2 and UO2 s.s. proceeded slowly at the interface between the solid and seawater. The congruent dissolution of Cs, I, and Ba with hexavalent uranium suggests that these typical elements might be released from the oxidized melted core even after decreasing the temperature of the debris. Pentavalent neptunium-239, which is the parent nuclide of Pu-239, exhibited a similar behavior to that of Cs, I, and Ba. The dissolution of Np in the simulated fuel debris of UO2 and UO2–ZrO2 was suppressed in the same manner as Zr(IV) and Ce(IV). The static leaching of Zr(IV) and Ce(IV) was not significant even from the oxidized uranium matrix. The leaching of hexavalent uranium might not facilitate the dissolution of tetravalent ions at the surface of fuel debris. Hence, the atmospheric conditions at the time of the formation of UO2–ZrO2 fuel debris should be critical to the leaching behavior of uranium, FPs, and minor actinides into the cooling seawater. A higher uranium concentration in the seawater samples as compared to the Milli-Q system might be attributed to the different contents of dissolved minerals, such as oxidants and microorganisms. Further experimental analysis of the contents of seawater and the chemical properties of neutron-irradiated solid solution samples is required in order to discuss the leaching mechanism in more detail.
Acknowledgement
This work was supported by the JSPS Grant-in-Aid for Scientific Research (S) [grant number 24226021]. The authors acknowledge Drs Toshiyuki Fujii, Akihiro Uehara, and the facility staff at the Research Reactor Institute, Kyoto University, for their great contributions to the experiment.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Hofmann P. Current knowledge on core degradation phenomena: a review. J Nucl Mater. 1999;270:194–211.
- Tanaka K, Miwa S, Sato I, et al. Effects of interaction between molten zircaloy and irradiated MOX fuel on the fission product release behavior. J Nucl Sci Technol. 2014;51:876–885.
- Nishihara K, Yamagishi I, Yasuda K, et al. Radionuclide release to stagnant water in the Fukushima-1 nuclear power plant. J Nucl Sci Technol. 2015;52:301–307.
- Tsukada T, Uozumi K, Hijikata T, et al. Early construction and operation of the highly contaminated water treatment system in Fukushima Daiichi Nuclear Power Station (I) – ion exchange properties of KURION herschelite in simulating contaminated water. J Nucl Sci Technol. 2014;51:886–893.
- Hijikata T, Uozumi K, Tsukada T, et al. Early construction and operation of the highly contaminated water treatment system in Fukushima Daiichi Nuclear Power Station (II) – dynamic characteristics of KURION media for Cs removal in simulated contaminated water. J Nucl Sci Technol. 2014;51:894–905.
- Collins ED, Campbell DO, King LJ, et al. A.I.Ch.E. Syrup. Series, 1982;213(78):9–15.
- Buckingham JS. Laboratory evaluation of zeolite material for removing radioactive cesium from alkaline waste solutions. Hanford (WA): United States Atomic Energy Commission; 1970. (Report ARH-SA-49).
- Choppin GR, Khankhasayev MK. Chemical separation technologies and related methods of nuclear waste management: applications, problems, and research needs (NATO Science Partnership Sub-Series 2). New York: Springer; 1999.
- Prevost T, Blase M, Paillard H, et al. Areva's Actiflo™-Rad water treatment system for the Fukushima Nuclear Power Plant. ATW-Int J Nucl Power. 2012;5:308–313.
- Katayama YB, Bradley DJ, Harvey CO. Status report on LWR spent fuel IAEA leach tests, PNL-3173. Richland (WA): Pacific Northwest Laboratory; 1980.
- Shoesmith DW. Fuel corrosion processes under waste disposal conditions. J Nucl Mater. 2000;282:1–31.
- Quinones J, Grambow B, Loida A, Geckeis H. Co-precipitation phenomena during spent fuel dissolution. 1. Experimental procedure and initial results on trivalent ion behavior. J Nucl Mater. 1996;238:38–43.
- Serrano-Purroy D, Clarens F, González-Robles E, Glatz JP, Wegen DH, de Pablo J, Casas I, Giménez J, Martínez-Esparza A. Instant release fraction and matrix release of high burn-up UO2 spent nuclear fuel: effect of high burn-up structure and leaching solution composition. J Nucl Mater. 2012;427:249–258.
- Sasaki T, Takeno Y, Kirishima A, Sato N. Leaching test of gamma-emitting Cs, Ru, Zr, and U from neutron-irradiated UO2/ZrO2 solid solutions in non-filtered surface seawater. J Nucl Sci Technol. 2015;52:147–151.
- Cohen I, Schaner BE. A metallographic and X-ray study of the UO2–ZrO2 system. J Nucl Mater. 1963;9:18–52.
- Kirishima A, Hirano M, Sasaki T, Sato N. Leaching of actinide elements from simulated fuel debris to seawater. J Nucl Sci Technol. Forthcoming 2015. doi:10.1080/00223131.2015.1017545
- National Astronomical Observatory of Japan. Chronological science tables 2005. Tokyo: Maruzen; 2004. Japanese.
- Belgrano A, Dunne JA, Bascompte J. Encyclopedia of ocean sciences. Vol. 2. Amsterdam: Elsevier; 2001. pp. 840–845.
- Sasaki T, Kobayashi T, Takagi I, et al. Hydrolysis constant and coordination geometry of zirconium(IV). J Nucl Sci Technol. 2008;45:735–739.
- Bard AJ, Parsons R, Jordan J, editors. Standard potentials in aqueous solution. New York: Marcel Dekker; 1985.
- Secretariat of the Team for the Decommissioning of TEPCO's Fukushima Daiichi Nuclear Power Station. Progress status and future challenges of the mid- and- long-term roadmap towards the decommissioning of TEPCO's Fukushima Daiichi Nuclear Power Station Units 1-4. Tokyo: Japanese Ministry of Economy, Trade and Industry (METI); 2013. Japanese. Available from: http://www.meti.go.jp/earthquake/nuclear/pdf/131128/ 131128_01ss.pdf
- Secretariat of the Team for the Decommissioning of TEPCO's Fukushima Daiichi Nuclear Power Station. Progress status and future challenges of the mid- and- long-term roadmap towards the decommissioning of TEPCO's Fukushima Daiichi Nuclear Power Station Units 1-4. Tokyo: Japanese Ministry of Economy, Trade and Industry (METI); 2015. Available from: http://www.meti.go.jp/english/earthquake/nuclear/ decommissioning/index.html#progress_status