

ABSTRACT
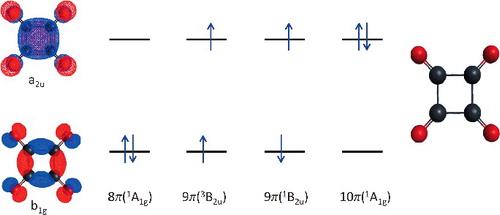
The four, closely spaced, lowest energy electronic states of the challenging, D4h-symmetric, 1,2,3,4-cyclobutanetetraone (C4O4) molecule have been investigated using high-level ab initio methods. The calculated states include the closed-shell singlet 8π(1A1g) state, the singlet 10π(1A1g) state, in which the π-type lowest unoccupied molecular orbital (LUMO) of the 8π(1A1g) reference is doubly occupied and the σ-type highest occupied molecular orbital (HOMO) is empty, and the open-shell singlet and triplet states, designated as 9π(1B2u) and 9π(3B2u), respectively, originating from single occupancy of the HOMO and LUMO. Our focus is on single-reference coupled-cluster (CC) approaches capable of handling electronic near-degeneracies in diradicals, especially the completely renormalised CR-CC(2,3) and active-space CCSDt methods, along with their CCSD and EOMCCSD counterparts. The internally contracted multi-reference configuration interaction calculations with a quasi-degenerate Davidson correction are performed as well. Our computations demonstrate that the state ordering is 9π(3B2u) < 8π(1A1g) < 9π(1B2u) < 10π(1A1g) and that the 8π(1A1g) − 9π(3B2u) gap is in the 7–11 kJ/mol range, in reasonable agreement with the negative ion photoelectron spectroscopy measurements, which give 6.27 ± 0.5 kJ/mol. In addition to the theory level used, geometry relaxation and basis set play a significant role in determining the state ordering and energy spacings. In particular, it is unsafe to use lower level, non-CC geometries and smaller basis sets.
GRAPHICAL ABSTRACT
Disclosure statement
No potential conflict of interest was reported by the authors.