

ABSTRACT
State-of-the art calculations have been performed for bimetallic transition metal clusters such as Pd19M19 (M=Co and Ni) by employing the linear combination of Gaussian-type orbitals density functional theory (LCGTO-DFT) approach. Structures and energy properties were calculated for these clusters. For each cluster, several dozens of isomers were studied to determine the lowest energy structures. Initial structures for the geometry optimisation were taken along Born–Oppenheimer molecular dynamics (BOMD) trajectories, considering several spin multiplicities. All structures were fully optimised without any symmetry restriction. The optimised structures were characterised by vibrational analysis. Lowest energy structures, relative stability energy, harmonic frequencies, binding energies, vertical ionisation potential, vertical electron affinity and spin density plots are reported. The obtained results are compared with data from the literature. The ground-state structure topology is the same for both clusters. The Pd atoms decorate the surface of the core formed by either Co or Ni atoms. This work demonstrates the importance of using ab initio BOMD simulations to fully explore the potential energy surface of large transition metal clusters.
GRAPHICAL ABSTRACT
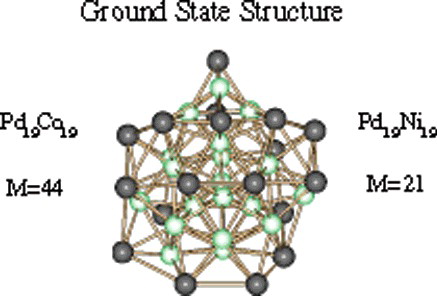
Structure and spin multiplicity of the ground state structure of Pd19M19 (M=Co, Ni) clusters. Dark grey spheres correspond to Pd atoms and light green spheres correspond to either Co or Ni atoms, respectively.
Acknowledgements
H. Cruz-Martínez acknowledges CONACYT for the doctoral fellowship 362306. The authors would like to thank Dr H. Arslan for providing them the Cartesian coordinates of the lowest energy structure of the lowest energy Pd19Co19 cluster reported in [Citation19]. The authors acknowledge the CGSTIC at CINVESTAV for providing HPC resources on the Hybrid Cluster Supercomputer ‘Xiuhcoatl’.
Disclosure statement
No potential conflict of interest was reported by the authors.