

GRAPHICAL ABSTRACT
ABSTRACT
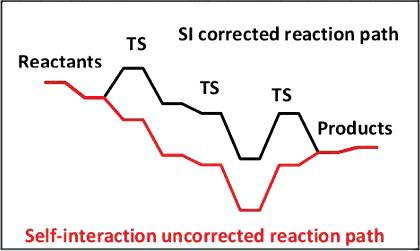
It is shown that modelling a catalytic reaction involving an atomic anion, such as iodide, may lead to unrealistic reaction paths because of the underestimation of the anion energy. The self-interaction error, at the origin of this feature is enhanced by the fact that electronic delocalisation, usually overestimated in DFT calculations, cannot occur in a single atom. The quantification of this error is approached by a linear transit calculation, from a transition state towards separate fragments. This is an efficient way to correct this error for GGA functionals. Interestingly the self-interaction error is shown to be less dependent of the Hartree–Fock exchange amount in hybrid functionals than expected. Unexpectedly, no significant improvement with long-range corrected functionals is obtained.
Acknowledgments
It is a pleasure for H.C. to dedicate this article to Andreas Savin, who has sown bright ideas in the contemporary development of DFT. The authors gratefully acknowledge the GENCI/CINES for HPC resources/computer time (Project cpt2130).
Disclosure statement
No potential conflict of interest was reported by the authors.