

ABSTRACT
We report calculations on the q(Yb) electric field gradient (EFG) for the X2Σ+ and A2Π1/2 electronic states of the ytterbium monofluoride (YbF) molecule at the molecular mean-field Dirac–Coulomb–Gaunt as well as scalar-relativistic coupled-cluster levels of theory using large uncontracted basis sets. Vibrational contributions are included in the final results. Our estimated nuclear quadrupole coupling constants of –3386(78) MHz and –2083(153) MHz for the X2Σ+ and A2Π1/2 states of 173YbF are in stark contrast to the only available experimental results (–2050(170) MHz and –1090(160) MHz) respectively, where the only similarity is the difference between the two values. Perturbative triple contributions in the coupled cluster treatment are significant and point towards the necessity to go to higher order in the coupled-cluster treatment in future calculations. We also present density functional calculations which show rather large variations for the Yb EFG with different functionals used; the best result was obtained using the CAM-B3LYP* functional.
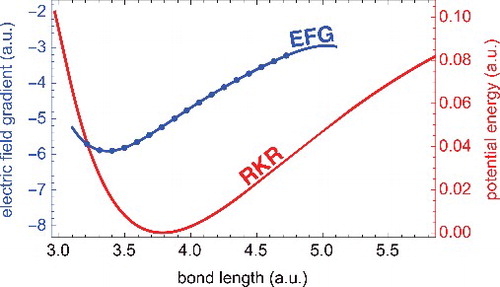
GRAPHICAL ABSTRACT
Acknowledgments
R.J.M. is grateful for research support provided by the Pomona College Sontag Fellowship Program as well as the Deutscher Akademischer Austauschdienst (DAAD). Part of the calculations were performed in the Computing Centre of the Slovak Academy of Sciences using the supercomputing infrastructure acquired in project ITMS 26230120002 and 26210120002 (Slovak infrastructure for high-performance computing) supported by the Research & Development Operational Programme funded by the ERDF.
Disclosure statement
No potential conflict of interest was reported by the authors.