

ABSTRACT
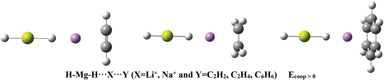
In the present study, H-Mg-H···X···Y (X = Li+, Na+ and Y = C2H2, C2H4, C6H6) triads have been investigated at MP2/6-311++G(2d,2p) computational level to characterise cooperative effects between hydride bonding and cation–π interactions. Molecular geometries, binding energies, cooperative energies and many-body interaction energies were evaluated. The diminutive energy values in triads with Li+ are larger than respective values in triads with Na+. The electronic properties of the complexes are analysed using parameters derived from the quantum theory of atoms in molecules methodology.
Disclosure statement
No potential conflict of interest was reported by the authors.