

ABSTRACT
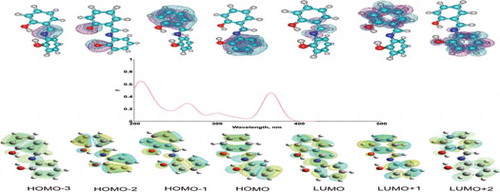
The time-dependent density functional theory (TDDFT) was applied in conjunction with the natural bond orbital analysis to examine the UV-Vis properties of 10 phenolic Schiff bases. The calculations were performed with different functionals, but main discussion refers to results obtained at the B3LYP/6-311+G(d,p) level of theory. The approach based on the natural localised molecular orbital clusters indicates similar behaviour for majority of examined compounds. The HOMO (“highest occupied molecular orbital”) cluster is delocalised over the ring which is electron richer, the HOMO-1 cluster is spread over the other ring, whereas the LUMO (“lowest unoccupied molecular orbital”) cluster is situated on the imino group. The two bands at long wavelengths correspond to the HOMO → LUMO and HOMO-1 → LUMO transitions, i.e. from both A and B rings to the imino group. The next band originates from a transition from the imino group to the imino group. The band at the smallest wavelengths originates from a transition from the A ring to the A ring, or from the B ring to the B ring. Our findings are in very good agreement with the existing literature data.
Acknowledgments
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (projects No 172016, 174028). The authors are very grateful to the research group of Professor Svetlana Marković for useful advice regarding this work.
Disclosure statement
No potential conflict of interest was reported by the authors.