

ABSTRACT
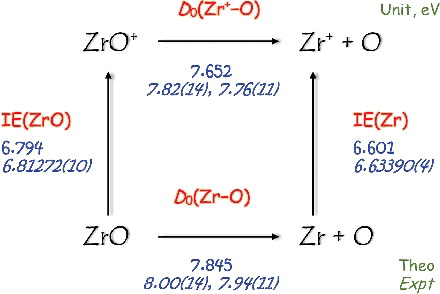
The ionisation energy (IE) of ZrO, the bond dissociation energies (D 0s) and the heats of formation at 0 K (ΔH ○ 0K f ) and 298 K (ΔH ○ 298K f ) for ZrO/ZrO+ are predicted by the coupled cluster methods utilizing upto single, double, triple and quadruple excitations and complete basis set limit approximation (CCSDTQ/CBS). The CCSDTQ/CBS approach also includes the zero-point vibrational energy, high-order correlation, core-valence (CV) electronic and spin-orbit coupling corrections while the scalar relativistic contribution is handled by employing pseudopotential basis sets. The present calculations yield IE(ZrO) = 6.794 eV and D 0(Zr+–O) − D 0(Zr–O) = −0.193 eV which are in good agreement with the respective experimental values of 6.81272(10) eV and −0.1788(1) eV determined in a two-colour laser-pulsed field ionisation-photoelectron study. The CCSD(T) and multireference configuration interaction (MRCI) methods with CV correlations included give equally remarkable predictions of the harmonic frequencies and the bond lengths for ZrO/ZrO+. This study together with the previous investigations has demonstrated that, with pseudopotential basis sets, the CCSDTQ/CBS protocol can be readily extended to investigate 4d-transition metal-containing diatomic molecules to yield comparable accuracy (±20 meV) to that achieved in the IE and D 0 predictions of 3d-transition metal-containing species.
GRAPHICAL ABSTRACT
Acknowledgments
This material is based upon the work supported by the National Science Foundation under grant CHE-1462172 and CHE-1642172. Cheuk-Yiu Ng is also grateful to Dr Huie Tarng Liou for his generous donation of research support for the Ng Laboratory. Kai-Chung Lau acknowledges the support from the Strategic Research Grant of City University of Hong Kong (Project No. 7004819).
Disclosure statement
No potential conflict of interest was reported by the authors.