

ABSTRACT
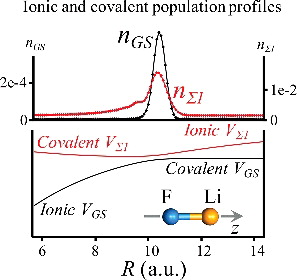
Rapid optical excitation of a molecule produces a nonstationary state localised in the Franck–Condon region. To move out of that region, one needs to propagate both the electronic and the nuclear state. We formulate the motion on a grid of nuclear coordinate. The coupling to the electric field is fully included in the Hamiltonian used for propagation. We use perturbation theory to analyse the results of dynamics from one grid point to another. The nonadiabatic coupling terms arise from propagating the electronic states. We apply the formalism to the simple case of a diatomic molecule in an approximate but accurate scheme that allows performing computation on a limited number of grid points. As the coherent dynamics unfolds, we expand the grid in the direction of the wave packet motion with the quantum chemical calculations of the electronic structure performed ‘on the fly’. The LiF molecule excited by a one-cycle IR pulse is used as a computational example. The 30-fs propagation through the crossing of the ionic and covalent states is overall adiabatic. The role of electron–nuclear coherences is emphasised.
GRAPHICAL ABSTRACT
Acknowledgment
We thank U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES). FR thanks the FRS-FNRS for its support.
Disclosure statement
No potential conflict of interest was reported by the authors.