

ABSTRACT
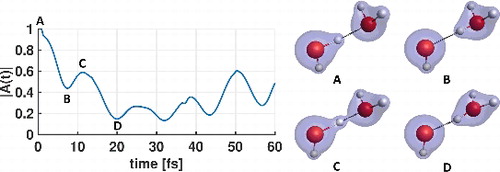
We introduce a new approach for analysing changes in electronic structure in the course of ab initio molecular dynamics simulations. The analysis is based on the time autocorrelation function of the many-body electronic wave-function. The approach facilitates the interpretation of dynamical events that may not be easily revealed by consideration of nuclear configurations alone. We apply the method to several illustrative examples: the shared proton vibration in the F−(H2O) complex, representing changes in strength of non-covalent interactions; proton transfer in the water dimer cation, as an example for chemical reactions in weakly bound systems; and the intramolecular proton transfer in malonaldehyde. In all cases, we observe distinct features in the time autocorrelation function when chemical changes occur. The autocorrelation function serves as an effective reaction coordinate, incorporating all degrees of freedom, including electronic ones. The method is also sensitive to changes in the electronic wave-function not accompanied by significant nuclear motions.
GRAPHICAL ABSTRACT
Acknowledgments
B. Hirshberg is supported through the Adams Fellowship of the Israel Academy of Sciences and Humanities. This work has been supported in Los Angeles by the Army Research Office through grant W911NF-16-1-0232 and the Alexander von Humboldt Foundation (Bessel Award to Anna I. Krylov). We thank Prof. Samer Gozem (Georgia State University) for his help with the ezDyson code.
Disclosure statement
AIK is a board member and a part owner of Q-Chem, Inc.