
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The structures and the vibrational dynamics of the complexes HHe are investigated experimentally (via mass spectrometry (MS)) and at high levels of electronic-structure theory. The MS measurements reveal interesting trends about the stability of the starting members of the HHe
family. The computations establish that the basically linear, strongly bound, symmetric triatomic molecular ion He(H
)He, with an equilibrium H–He distance of 0.925 Å and about 2/3 but at least 1/2 of the positive charge on H, is the molecular core of all of the
complexes. Definitive quantum-chemical results are obtained for HHe
and HHe
, including the proton affinity of He (computed to be
cm−1 via the focal-point analysis (FPA) scheme), the FPA isomerisation energy between the two linear isomers of HHe
(
cm−1), and the dissociation energy of the HHe
HHe
+ He reaction, with an FPA estimate of
cm−1. The structural isomers of the He-solvated complexes are discussed up to n=18. A useful notation, [k−l−m]-HHe
, is introduced to characterise qualitatively the three possible belts around the He–H
–He core in HHe
(
), where l denotes the number of He atoms in the central belt and
denote the number of He atoms in the top and bottom belts. Capping He atoms attached to the belts can be indicated by sub- and superscripts. Several possible indicators of microscopic superfluidity are investigated: He evaporation energies, rotational constants, and vibrational fundamentals.
GRAPHICAL ABSTRACT
1. Introduction
Doped He droplets, both with fermionic 3He and bosonic 4He, have become standard media for high-resolution molecular spectroscopy experiments [Citation1–4], they were termed ‘the ultimate spectroscopic matrix’ [Citation5]. Of particular relevance for the present study, spectroscopic investigations of doped He nanodroplets of widely different size yielded an extremely interesting property: microscopic superfluidity (MSF) [Citation4,Citation6–18]. One of the aims of MSF studies has been the establishment and the detailed characterisation of the evolution of superfluidity, starting from small (van der Waals) clusters, showing of course no sign of MSF and involving just a couple of He atoms, up to thousands if not hundreds of thousands of He atoms, showing pronounced superfluidity at the extremely low temperatures (around and in cases significantly below 1 K) of the experiments. A surprising finding of experimental MSF studies has been the establishment of the fact that clusters formed by a relatively small number of He atoms, on the order of 10-100, surrounding neutral molecules like OCS [Citation11,Citation14], NO [Citation17], and HCCCN [Citation18], display signs of superfluidity. Experimental signatures of MSF include (a) the increased moments of inertia and the disappearance of the Q branch in the spectra of He-solvated linear dopant molecules [Citation19], and (b) characteristic change in the effective n-dependent rotational constants, where n is the number of solvating He atoms, associated with free rotation of the microsolvated dopant within the cluster. Ground-state quantum Monte Carlo [Citation14], reptation quantum Monte Carlo [Citation18], finite-temperature path-integral Monte Carlo (PIMC) [Citation17], and two-fluid hydrodynamic [Citation20] model studies also provided evidence supporting the superfluidity-based interpretation of the limited experimental observations.
Microsolvation studies could involve, besides 3He and 4He, molecular hydrogen, especially para-H. We are aware of only a small number of studies utilising para-H
for investigating MSF [Citation19,Citation21,Citation22].
Experimental spectroscopic microsolvation studies, whether performed in the microwave (MW) or the optical (infrared, IR) regions, face several considerable challenges. One difficulty has always been the separation of clusters of different size. If He solvates not neutral molecules but ions [Citation23–31], this difficulty could be circumvented to some extent and the dynamics and the resulting spectra of microsolvated ions could be studied experimentally using ion trap techniques [Citation32]. The perhaps simplest family of microsolvated cations, which has many potential interests and is especially relevant for astronomy, has H, H
, and H
as potential dopants.
Despite the fact that the HHe
molecular ions and their He-solvated complexes are formed by the two most abundant elements of our known universe, surprisingly little is known about the structure, energetics, and especially the nuclear dynamics of H
He
species. Most of the experimental investigations, especially for larger x and (in particular) larger n values, are mass spectrometry (MS) studies [Citation33–39]. MS does provide important information about the existence of H
He
species (vide infra), and even some limited information about their relative stabilities. In this study we supplement the available experimental MS results with an eye on future high-resolution spectroscopic studies on some H
He
systems (see Section 2). Nevertheless, detailed structural and especially dynamic information cannot be obtained this way. Thus, it is unfortunate that there are only a handful of spectroscopic and computational investigations on the smaller members of the H
He
family [Citation36,Citation40–69], and even they sometimes contain data less well understood. High-resolution spectroscopic [Citation41,Citation43–46,Citation48,Citation49,Citation52,Citation53,Citation62,Citation66] and first-principles quantum chemical information [Citation40,Citation42,Citation47,Citation51,Citation57,Citation58,Citation60,Citation61,Citation63–65] is available mostly for the different isotopologues of the HHe
molecular ion, the simplest polar molecule and a favourite system of few-body quantum-mechanical studies.
Apart from some early confusion, all electronic-structure computations agree that HHe is a highly stable molecular ion and it forms the core, the ‘dopant’, the ‘chromophore’, of He-solvated HHe
clusters. This is in contrast to other solvated ions which do not exhibit this structural motif, as, e.g. LiHe
complexes [Citation31]. There have been a few attempts to compute, at various levels of electronic-structure theory, optimised equilibrium structures and dependable relative energies for HHe
for low values of n but larger than n=2 [Citation36,Citation54–58,Citation70]. For example, in 2000, Balta and Gianturco [Citation57] performed single and double excitation configuration interaction (MRD-CI) computations with the compact cc-pVTZ basis of Dunning [Citation71] and obtained two minima for HHe
: a T-shaped and a linear one. As detailed below, our more extensive computations presented in this paper, performed up to single, double, triple, and quadruple excitations within the coupled-cluster (CC) theory, named CCSDTQ, and with basis sets up to aug-cc-pV6Z [Citation72], clearly indicate that while the T-shaped form is indeed a minimum, the linear form becomes a second-order transition state when basis sets more extensive than aug-cc-pVTZ are employed. In another computational study, Filippone and Gianturco [Citation54] indicated breaking of the ‘ideal’ point-group symmetry of
and
for the equilibrium structures of the He-solvated complexes HHe
and HHe
, respectively. For HHe
, the two He atoms solvating H
of the HHe
core are placed at a distance of 2.30 and 2.14Å, yielding a strongly asymmetric planar structure. As to HHe
, the H
–He distances are about equal, 2.20Å, but they are tilted toward one side of the complex and the equilibrium geometry, due to the attraction of the He atoms, does not display the fully symmetric
point-group symmetry. Balta, Gianturco, and Paesani [Citation58] came up with evaporation energy estimates of HHe
complexes up to n=6. The computed evaporation energies show a steady decreasing tendency, changing from 0.7 to 0.4 kcal mol −1 when going from n=3 to n=6. This computational result is in contrast to the results of experimental MS studies [Citation35,Citation39] showing that n=6 is a ‘magic number’ and HHe
has pronounced stability (see also our own MS study in Section 2 supporting this statement). Classical and quantum dynamical studies have also been attempted on these systems [Citation73].
In this paper, after reporting relevant MS results obtained for small x and n values, we make an attempt to explore the configuration space of a large number of HHe species, up to n=18, and locate at least the lowest-energy isomers. Note that for at least some of these species one can easily get trapped in the web of polytopism [Citation74–76], as the level of electron correlation and the size of the basis set influence the computed results, sometimes even in a qualitative way. Therefore, (a) some previous studies suffered from the use of relatively low levels of wave-function-based electronic-structure theory, and (b) density-functional-theory (DFT) results should mostly be viewed with caution for this class of molecules, although specialised parametrised techniques [Citation77–81] seem to provide outstanding results.
2. Mass-spectrometry results
As mentioned in the Introduction, a couple of advanced experimental MS studies [Citation34,Citation35,Citation39] have been performed on HHe ions. These studies were designed to explore the generation and the stability of the HHe
ions produced under various experimental conditions.
Following a pioneering study of Veatch and Oskam [Citation34], Kojima et al. [Citation35] injected H and H
into a drift tube filled with He at a temperature of 4.4 K and generated HHe
ions with n up to 14. Through the measurement of the drift-field dependence of the ion yield, they concluded that the n=13 (and possibly also the n=6) cations are particularly stable members of the HHe
family. In their words [Citation35], ‘HHe
is relatively more stable than other clusters’ and ‘the 14th He atom is bound much less tightly than the last He of HHe
’. Unfortunately, based on the limited data available to them, Kojima et al. [Citation35] considered HHe
as the chromophore of the HHe
complexes, while we know today that the chromophore is HHe
. Thus, their suggestion that ‘HHe
has an HHe
-centred icosahedral structure’ turns out to be incorrect. In 2013, Bartl et al. [Citation39] obtained high-resolution mass spectra (
) of helium nanodroplets which have been doped with H
or D
and subsequently ionised. The very high resolution of the spectrometer used enabled them to distinguish the ionic complexes with masses up to 120 u, corresponding to almost 30 He atoms. Bartl et al. [Citation39] were able to confirm the results of Kojima et al. [Citation35], most importantly that n=6 appears to be a magic number for the HHe
species. The ion yield obtained by Bartl et al. [Citation39] also showed an abrupt drop from n=13 to 14. They associated this drop with a ‘similarly abrupt drop in the evaporation energy’ [Citation39]. It is worth adding that the ion-yield figure (Figure 2a of Ref. [Citation39]) suggests ‘no further magic numbers, at least not below n=30’.
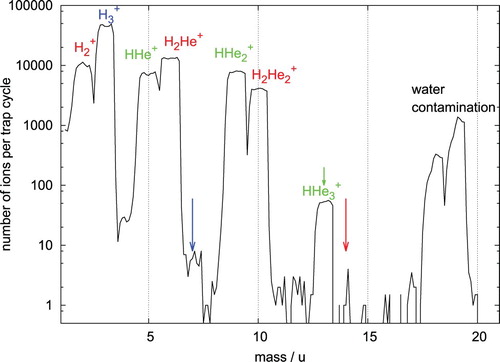
To confirm and extend these results, we performed MS experiments applying the cryogenic 22-pole trap apparatus COLTRAP [Citation82,Citation83]. Similar to the drift-tube experiments [Citation35], we injected hydrogen cations into a cold environment containing He gas, but at much lower kinetic and internal energies. During the first set of experiments, H ions, generated by ionising a low-density (H
:He = 1:4) gas mixture in a storage ion source, were mass selected in a quadrupole mass filter, and then injected with low energy (typically less than 1 eV) into the 4 K cold 22-pole ion trap. A strong helium gas pulse at the beginning of the trapping cycle leads to the mass spectrum shown in Figure , which was recorded after the trapping time of 1.5 s. A rich variety of H
He
ions can be observed in Figure . Apart from the tagging of the injected H
, leading to the formation of H
He
ions (n=1,2 and 3, shown in red in Figure ), H
ions (shown in blue) are also produced in large quantities. This is due to a small H
contamination traversing the trap (leaking from the ion source into the trap) and the fast exothermic reaction H
+ H
H
+ H. The blue and red arrows in Figure show a strong drop of many orders of magnitude in the number of the complexes H
He
and H
He
and indicate special stability for H
He
and H
. The reluctance of H
to attach a helium atom (as compared to H
) can be rationalised by the fact that this is a closed shell species and even the first He atom ‘solvating’ H
is only loosely attached to the ion. The ternary rate coefficient for the attachment of He to H
at 3.7 K has been given by Savić et al. [Citation67].
Figure 1. Mass spectrum recorded after trapping mass-selected H ions in a cold (4 K) 22-pole ion trap for 1.5 s. At the beginning of the trapping cycle, a strong and short helium pulse (
cm−3,
ms) has been admitted to the trap. The resulting ion masses are explained in Section 2 of the text. The arrows indicate a relatively low probability to attach a further He atom to H
(blue), HHe
(green), and H
He
(red).
HHe species, forming the topic of this paper, are also readily produced in the trap. These ions stem from vibrationally excited H
(v) generated in the ion source (the energy of the ionising electrons is 17 eV). When entering the trap, the otherwise endothermic reaction H
(v) + He
HHe
+ H is enhanced for
[Citation84]. Further tagging by He atoms leads to the HHe
species shown in green in Figure . Again, a drop of two orders of magnitude can be seen there for n=3 (green arrow), corroborating the special stability of the linear ion HHe
. This extra stability of HHe
can be seen in the mass spectra of Kojima et al. [Citation35], as well (see their Figure 1), though it is not explicitly mentioned there, because HHe
was assumed to be the chromophore in that paper. To optimise the experimental conditions for generating HHe
ions, we performed further test measurements, injecting a mass-selected ensemble of HHe
ions (m=5 u), generated similarly by a helium-hydrogen mixture in the ion source, into the 4 K cold ion trap. Our optimisation attempts led to the mass spectrum shown in Figure . The tagging of the injected HHe
in a constant cloud of He atoms leads to HHe
and HHe
ions, as well as higher complexes, and features special stability (usually associated with shell closure) not only for n=2 but also for n=6. Unfortunately, HHe
complexes with about
are difficult to investigate in the cold ion trap experiment in its present configuration (see Figure ); therefore, the reported stability for n=13 could not be confirmed.
Figure 2. Mass spectrum recorded after injecting mass-selected HHe ions into a 4 K cold 22-pole ion trap. The initial mass-selected ion bunch with m=5 u is drawn in thick green. The trap is constantly filled with helium (
cm−3). The trap time is 0.8 s. Ternary attachment of helium leads to the observed HHe
ions with prominent, order-of-magnitude drops in the number of ions observed after n=2 and n=6, indicating special stability for those species. All other masses are due to small contaminations of H
or H
O co-admitted to the trap. The H
ions (m=3 u) stem from the proton hop reaction HHe
+ H
H
+ He.
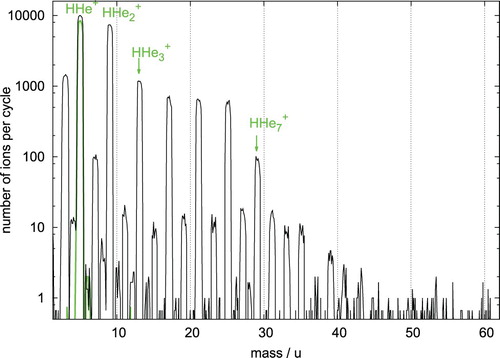
The fundamental nature of HHe makes it a promising target for future spectroscopic experiments. These experiments are made simpler by the following characteristics of HHe
: (a) it has a singlet ground electronic state, in contrast to, for example, H
He
and its He-solvated clusters; (b) it has a linear equilibrium structure; and (c) it has prominent stability. Most importantly, the weakly bound He atom in HHe
(green arrow in Figure ) opens up the possibility to perform high-resolution rovibrational spectroscopy experiments applying the method of state-dependent attachment of He atoms. This method is based on the fact that rovibrational excitation of a cation lowers the probability of attaching He atoms in a ternary collision process at 4 K. For instance, this method has been successfully employed for the high-resolution rovibrational spectroscopy of fundamental cations, such as CH
[Citation83,Citation85], O
H
[Citation86], and CH
[Citation87]. By counting the number of HHe
ions as a function of the frequency of the irradiating IR laser, it should be possible to record a rovibrational spectrum of HHe
in a future experiment. The IR-active anharmonic(harmonic) antisymmetric (
) stretching vibration of linear HHe
is a promising target, predicted to be at 1345(1539) cm−1 at the aug-cc-pVQZ MP2 level. With a suitable cw light source (most probably a quantum cascade laser) operating in that region, we intend to tackle this fundamental system in the Cologne laboratories. Even rotational spectroscopy is feasible with this action spectroscopic method [Citation87–90]. The computed rotational constant of HHe
, again at the aug-cc-pVQZ MP2 level, is
cm−1.
Characterization of the HHe species with
can be performed by conventional predissociation spectroscopy. For these species, the evaporation energy of the outer helium atom (Figure , vide infra), is well below the antisymmetric stretch excitation of the inner HHe
chromophore, leading to detectable dissociation upon resonant excitation. Using the wide tunability and the high power of the FELIX free-electron laser [Citation91], we have detected this vibrational feature in a preliminary measurement at about 1290 cm−1 for n=3−6 at low resolution (not shown here). It is our intention to refine these features with high-resolution lasers in rovibrational detail. In combination with rotational excitation, a recently developed double-resonance technique [Citation92,Citation93] can also be applied to record high-resolution rotational spectra of such weakly bound species. By recording the rotational spectra of HHe
for increasing n, signatures of microscopic superfluidity may become detectable.
3. Theoretical and computational details
Our computational investigation of the structure and energetics of the smaller members (n=1 and 2) of the closed-shell HHe family is based on the focal-point-analysis (FPA) technique [Citation94,Citation95]. This requires the execution of a large number of electronic-structure computations using various levels of electron-correlation treatment and basis sets and yields uncertainties for the computed quantities. For larger members of the family, the detailed investigations had to be restricted mostly to the aug-cc-pVTZ and aug-cc-pVQZ MP2 levels.
3.1. Electronic structure computations
In this study the correlation-consistent (cc) family [Citation71] of atom-centred, fixed-exponent, Gaussian basis functions has been employed for the electronic-structure computations. Due to the nature of the cations, especially taking into account the noble-gas nature of He and the large HeHe distances, diffuse (‘aug’) functions have been part of all basis sets used. The aug-cc-pVXZ basis sets with X=3(T), 4(Q), 5, and 6 have been used, occasionally abbreviated as aXZ.
The MP2 computations have been carried out with the Gaussian16 (G16) program package [Citation96]. All CC electronic-structure computations utilised either the CFOUR [Citation97] or the MRCC [Citation98–100] packages. To obtain proper results, especially harmonic frequencies, it proved to be essential to increase the standard convergence criteria of CFOUR by almost two orders of magnitude. Vibrational anharmonicities have been computed with the help of the G16 package [Citation96].
3.2. Breaking of the assumed symmetry of equilibrium structures
In several of the He-solvated structures the naively expected point-group symmetry of the equilibrium structure is ‘lowered’ by the actual geometry optimizations. In a few cases stationary points which are minima at a given level of electronic-structure theory turn out to be transition states at other levels. Such problems are not unknown in electronic structure theory, relevant examples include the polytopic LiCN [Citation75], SiC [Citation76,Citation95], (CH)
[Citation74], (CH)
[Citation74], and (CH)
CO [Citation74] molecules. There can be two reasons behind these difficulties: (a) competition between the weak
,
, and
interactions (attractions) yields asymmetric distortions; and (b) inadequacy of or symmetry breaking in the (single-reference) electronic wave functions. The interactions noted are fully physical; thus, we investigated in detail the latter issues to rule out possible problems with the electronic-structure computations utilised during the present study.
The diagnostics [Citation101] of coupled-cluster theory, i.e. the Euclidian norm of the
amplitudes given by the CCSD procedure, is a common diagnostic for nondynamical electron correlation. It has been used in this study to investigate whether single-reference techniques would provide suitable results for the HHe
systems. The computed
values are small in all cases investigated; for example,
for HHe
. Thus, it appears to be safe to use single-reference coupled-cluster theory during this study.
Stability of the Hartree–Fock determinant was also investigated in several cases. In all cases studied no instability was indicated by the G16 stability analysis computations.
As to the competition between the H+He, HeHe, and He
He interactions, it is clear both from previously computed energy results and those of the present study that by far the largest energy gain is obtained if the solvating He atoms attach to the central proton core, which in all He-solvated complexes has a Mulliken charge larger than +0.5. The H
He interaction energy appears to be about 300 cm−1 at equilibrium. The second most important interaction is that of the He atoms of the ‘linear’ triatomic core, having a substantial positive Mulliken charge of about 0.1–0.3 (depending on the basis), with the solvating He atoms. The magnitude of this interaction energy is about 100 cm−1. The attractive He
He interaction is weak, on the order of a few cm−1 [Citation102], but it is responsible for the relative stability and the size of the three belts formed around the three positively charged atoms of the He(H
)He core of the He-solvated complexes (vide infra).
3.3. Geometry optimizations
The equilibrium structures of all HHe complexes have been determined at the aug-cc-pVTZ MP2 level. Geometry optimizations have been carried out also at the CCSD, CCSD(T), CCSDT, CCSDT(Q), and CCSDTQ levels with basis sets up to aug-cc-pV6Z for small n values. Note that CCSD and CCSDTQ correspond to full configuration interaction (FCI) for HHe
and HHe
, respectively.
The structures optimised at the aug-cc-pVQZ CCSD(T) level served as reference structures for the FPA analyses of this study. Since the bonding of He to the cationic core in the He-solvated cases is extremely weak, great care must be exercised when the geometry optimisation is stopped.
3.4. Focal-point analysis (FPA)
During the last 25 years the technique of focal-point analysis [Citation94,Citation95] has been employed a large number of times to study molecular energy differences as well as molecular potential energy surfaces (PES) [Citation103]. These studies include not only small [Citation76,Citation104–106] but also large systems, like peptide building blocks[Citation107] and amino acid complexes [Citation108]. Within the FPA scheme it is usual to extrapolate the energies and the energy increments to the complete basis set (CBS) limit [Citation95,Citation109,Citation110]. Most importantly, the FPA scheme allows the estimation of the uncertainty of the computed relative (interaction) energies [Citation111].
The auxiliary relativistic corrections determined in this study for each species were obtained at the mass-velocity and one-electron Darwin (MVD1) level [Citation112] employing the aug-cc-pVQZ basis and the CCSD(T) level of electronic structure theory. These relativistic estimates should be sufficiently accurate for the very light HHe systems even at the level of accuracy sought during this study.
The diagonal Born–Oppenheimer corrections (DBOC) [Citation113] were estimated at the aug-cc-pVQZ HF level.
3.5. Harmonic and anharmonic vibrational frequencies
To obtain highly accurate relative energies through the FPA approach, it is preferential to use anharmonic ZPVEs. Harmonic ZPVEs have been obtained during this study, mostly to check whether an optimised stationary point corresponds to a minimum or not. These harmonic ZPVE estimates are also reasonably accurate for the purpose of correcting relative electronic energies, as done in this study.
Next, we must discuss why we are not computing anharmonic perturbative corrections to the ZPVE for the large(r) He-solvated clusters. The reason is that they are expensive and, perhaps even more importantly, make limited sense once there is a large number of fundamentals below 100 cm−1, which is certainly the case for large(r) n values. One must take the lack of anharmonic corrections into account when deciding about the uncertainty of the vibrational corrections to the relative energies.
4. Molecular cations
The attachment of one and two He atoms to H results in relatively stable molecular cations with a linear equilibrium structure (see Figure ). While in Figure and subsequent figures of the structures of the HHe
cations the Mulliken charges obtained from aug-cc-pVTZ RHF densities are denoted on the atoms, they only serve qualitative purposes and should not be taken literally, especially since Mulliken charges are dependent on the level of electronic structure theory utilised for their computation. In HHe
the internuclear distance is 0.775 Å (a nonrelativistic, CBS FCI value), suggesting the presence of a relatively strong, ‘true’ chemical bond. As it turned out during the electronic-structure computations of this study, the next member of the HHe
series, the He(H
)He molecular cation, forms the core (the ‘dopant’) of all the solvated HHe
complexes whose equilibrium structure has been optimised. Thus, in this section the strongly-bound HHe
and HHe
species are treated, separate from the truly He-solvated cations, discussed in Section 6.
Figure 3. Equilibrium structures of HHe (left) and two isomers of HHe
(middle and right) with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, denoted on the atoms (H is white, He is light blue).
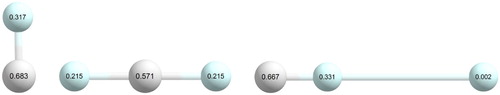
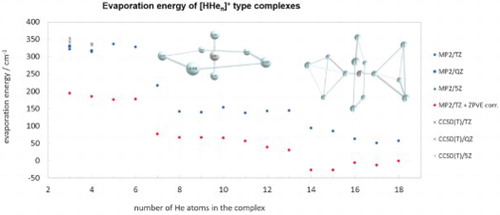
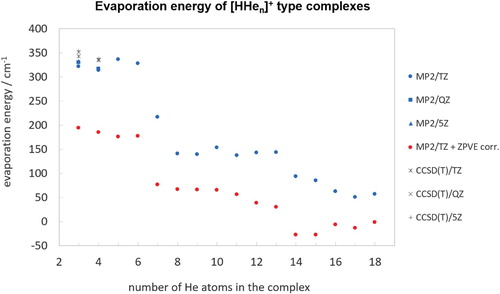
Figure 4. Evaporation energies of the HHe complexes (n=3−18) obtained during this study. Only the global minimum for each n is considered.
4.1. HHe
The molecular cation HHe, sometimes called the hydrohelium cation (see the left panel of Figure ), possibly the first molecule formed in space [Citation114] and the founding member of the HHe
series, was detected in a mass spectrum almost one hundred years ago [Citation33]. It is a molecule of considerable importance for the astrochemistry and astrophysics of certain stars [Citation115,Citation116]. Furthermore, the four-body HHe
molecular cation serves as a benchmark system for high-accuracy first-principles computations. These computations often go beyond the Born–Oppenheimer separation of nuclear and electronic motions and take into account non-adiabatic, relativistic, and quantum electrodynamic (QED) effects [Citation61]. Note also that Telmini and Jungen developed the so-called halfium model [Citation117], allowing the treatment of excited states of diatomic molecules with just two active electrons, and applied it to compute and characterise the Rydberg states of HHe
[Citation118].
The equilibrium H–He bond length, (HHe), is only 0.775 Å, corresponding to the nonrelativistic CBS FCI limit. This structure yields a large equilibrium rotational constant,
cm−1, while the corresponding harmonic frequency is
cm−1 (from here on the assumed uncertainties of some of the computed quantities are indicated, to comply with recent recommendations [Citation111] of the atomic and molecular physics community). The related data corrected for vibrational effects are
and
cm−1. The highly accurate experimentally determined
and
values of 4HeH
are 33.55867000(98) [Citation53] and 2910.958174(18) [Citation66] cm−1, respectively, in reasonable agreement with the computed results.
The harmonic zero-point energy estimate for HHe is
cm−1. The anharmonic correction to this value is
cm−1, yielding an anharmonic ZPVE estimate of
cm−1.
The simple reaction He + H
HeH
defines the proton affinity (PA) of the helium atom. As detailed in Table , the computed electronic PA of He, obtained as part of this study, is
cm−1. Note the very small electron-correlation contribution to this value. This value should be corrected with the anharmonic ZPVE of HHe
, as well as with the (relatively substantial) DBOC correction,
cm−1, and the minuscule relativistic correction,
cm−1. Overall, our FPA estimate of the proton affinity of the ground-state 4He atom is
cm−1. This computed PA of He compares extremely well with the available literature values, namely 14,873 [Citation42] and 14,863(8) [Citation119] cm−1.
Table 1. Focal-point-analysis table of the proton affinity of the helium atom, corresponding to the reaction .
4.2. HHe
The ground electronic state PES of HHe exhibits two linear minima. The global minimum corresponds to the symmetric He(H
)He arrangement with an equilibrium structure of
point-group symmetry and
(HHe) = 0.925 Å, corresponding to the nonrelativistic CBS FCI limit. This bond length is significantly longer, just as expected, than that in HeH
, 0.775 Å, showing the considerably weaker binding of He by the H
core within the symmetric HHe
ion. At the aug-cc-pVQZ MP2 level, the
rotational constant of linear HHe
is 2.370(2.473) cm−1. VPT2 theory provides the following anharmonic(harmonic) fundamentals at the aug-cc-pVQZ MP2 level: 882(959), 893(1140), and 1345(1539) cm−1, and the following anharmonic(harmonic) ZPVE values: 2265(2298) cm−1. The harmonic frequencies at the aug-cc-pV5Z FCI level are only slightly different at 958(
), 1138(
), and 1561(
) cm−1, yielding a harmonic ZPVE estimate of 2308 cm−1.
The secondary minimum of point-group symmetry, when the He atom attaches to the He end of the diatomic hydrohelium cation, corresponds to the He-solvated HHe
molecule. This stationary point is a minimum at all levels of theory studied. The distance between the two He atoms is 2.12 Å in this case (it is 1.85 Å for the global minimum). As can be seen in Figure , the solvating He atom acquires a very small positive charge, for the other two atoms the charge distribution of the secondary minimum resembles closely that of HHe
(where about 2/3 of the full charge is located on H and 1/3 on He). The FPA energy difference between the two isomers of HHe
is huge on the energy scale of cations containing only H
and He, it is
cm−1, without taking into account the ZPVEs (). The energy difference of the two linear isomers of HHe
becomes
cm−1 when the ZPVE correction is taken into account. Interestingly, electron correlation and zero-point vibrational contributions to the isomerisation energy basically cancel each other out (Table ).
The reaction energies of the HHe
HeH
He (Table ) and HHe
H
He (total atomisation energy (TAE), which can be deduced from Tables and by adding the appropriate entries) processes suggest that the binding of two He atoms to H
is a strongly exothermic process. The CBS-FCI-based FPA value for TAE of HHe
is
cm−1, compared to
cm−1 for HHe
. Convergence of the CC series is pronounced, the correction above CCSDT is as small as +1 cm−1 for the dissociation energy of HHe
. The ZPVE-corrected TAE is
cm−1.
Table 2. Focal-point-analysis table related to the dissociation energy of , .
Table 3. Focal-point-analysis table of the isomerisation energy related to the two linear isomers of (see Figure ).
Based on the pure electronic reaction energy of Table and the ZPVEs, our estimated dissociation energy of the HHe
HeH
He reaction is
cm−1. A substantial part of the uncertainty comes from the ZPVE correction.
The existence of two minima on the ground-state PES of HHe means that even this simple triatomic cation would exhibit interesting and complex rovibrational resonances at excitation energies close to the energy of the secondary minimum, which can be studied, for example, by high-resolution overtone spectroscopy in an ion trap. The experimental studies could be nicely complemented by fourth-age [Citation120] variational nuclear-motion studies if an accurate global PES was made available.
5. A notation recommended for He-solvated HHe () complexes
Before proceeding to the actual presentation of the structures of the HHe (
) complexes, provided in Section 6, a useful way to qualitatively characterise the structures of the HHe
complexes investigated in this study is given.
The three atoms of the He(H+)He core (the dopant in a He-solvated HHe cluster with
) have positive partial charges. The largest positive charge is always on the central atom but the two strongly-bound He atoms also acquire substantial charges. For the linear He(H
)He complex the Mulliken charges are +0.571 and +0.215 for the H and He atoms, respectively (see Figure ). The equilibrium structures of the larger He-solvated complexes reflect the easily explainable tendency that the larger positive charge an atom possesses the more He atoms solvate that atom of the core. Since He atoms prefer to solvate a dopant in a planar (or nearly planar) fashion (as a result of the different attractive interactions), we can have three ‘belts’ (‘rings’, ‘toroids’, ‘donuts’) in a He-solvated HHe
complex, placed more or less perpendicularly to the linear He(H
)He core. For the lowest-energy isomers the belt containing the largest number of directly-bonded He atoms is the central belt around H
and this belt is filled up first. The two other belts, the ‘upper’ (top) and the ‘lower’ (bottom) belts have less He atoms in the lowest-energy complexes. It is an interesting question whether symmetric or less symmetric arrangements are preferred for the top and bottom belts, and the answer is case specific. Nevertheless, the energy differences are exceedingly small and thus the dependability of the electronic-structure computations performed in this study can arguably be questioned for larger n values.
Due to the presence of three rings, a useful notation for He-solvated H clusters is the
notation, where l denotes the number of He atoms surrounding the central H
atom and k and m denote the number of He atoms in the top and bottom belts. It is useful to choose k to be larger than m.
In some of the complexes studied the outer belts contain capping He atoms. This bonding can be indicated by using upper indices in front of k and m when the capping He atom has more or less equal bonding to all the He atoms of the belt. If only selected atoms of the belts are solvated, the hapticity concept (the η-notation) [Citation121] of inorganic chemistry could be employed for the notation. Nevertheless, we chose to indicate these structural features using subscripts after k and m. This complication in the structure of the isomers of HHe is observed first for the n=13 case, when 11 He atoms solvate the He(H
)He core. If more than one He atom has
-type bonding in one of the belts, this can be indicated by repeated subscripts. For example,
means that a 4-membered belt (say, k=4) is solvated by two He atoms showing
-type bonding. The additional He atoms can share a common He atom (syn, s) or can take opposite sides (anti, a) of the 4-membered ring. These
-type bonding features can be indicated as part of the subscripts (this is an issue only for HHe
.) Capping He atoms can be indicated as superscripts.
6. He-solvated complexes of the He(H)He core
When the He(H)He core is solvated with He atoms, the He
H distances are (considerably) longer than 1.7 Å, while the He
He distances between the solvating He atoms and those of the core are even longer, at least 2.1 Å). While the structures of the He-solvated complexes are discussed in separate subsections, we present several tables summarising the main computational results of this study (Tables ).
Table 4. Rotational constants, in GHz, of the (n=1−18) species, computed at the aug-cc-pVTZ MP2 level. Only the global minimum for each n is considered.
Table 5. The four highest fundamentals, in cm−1, of the HHe (n=1−18) species, computed at the aug-cc-pVTZ MP2 level. Only the global minimum for each n is considered.
Table 6. Harmonic zero-point vibrational energies of the HHe isomers considered in this study.
Table 7. Successive electronic evaporation energies, in cm−1, defined through the reaction HHe HHe + He, of the HHe species considered in this study, where only the global minimum for each n is considered (note that consideration of the ZPVE changes the stability order of the two lowest-energy isomers of HHe, HHe, and HHe (see text for details)).
The equilibrium rotational constants of the HHe (n=1−18) species are collected in Table , obtained at the aug-cc-pVTZ MP2 level. Table contains the four highest fundamentals of the HHe
(n=2−18) species, computed at the aug-cc-pVTZ MP2 level, corresponding to the two bending and the two stretching vibrations of the He(H
)He core. Only the global minima have been considered when the data of Tables and have been assembled. Table contains the harmonic ZPVE values for all the complexes HHe
, n=1−18, obtained at several levels of electronic structure theory. Finally, the evaporation energies of all the complexes studied are listed in Table and depicted in Figure .
A couple of important questions are addressed in this and the following sections. First, we discuss the structure, the symmetry, and the harmonic vibrations of the complexes. Second, we look for the presence of solvation shells. Third, we investigate the role a pure quantum effect, the zero-point vibrational energy, has on the strength of the complexes, defined through the evaporation reaction HHe
HHe
He. Fourth, we look for tell-tale signatures of microscopic superfluidity.
6.1. HHe
In the simplest truly He-solvated complex of the HHe series the third He binds to H
carrying the largest partial positive charge (see Figure ). The structure of this He-solvated complex is thus [0–1–0]-HHe
. This is the only isomer we found on the PES of HHe
.
Figure 5. Equilibrium structures of HHe (left) and HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).

The polytopic nature of some of the HHe complexes shows up here in that (a) at the CCSD(T)/aug-cc-pVQZ level, and only there, the structure with a linear He(H
)He core is also a minimum, higher in energy by only 5.6 cm−1 than the slightly bent,
point-group-symmetry isomer, the ‘true’ equilibrium structure of HHe
; and (b) the linear arrangement of the four atoms, He(H
)He
He, is a minimum at the aug-cc-pVTZ MP2 and CCSD(T) levels, with very slightly unequal HHe distances within the core. When larger basis sets are used, either aug-cc-pVQZ or aug-cc-pV5Z, the linear structure becomes a second-order transition state, and the imaginary harmonic frequencies are larger in the case of the a5Z basis, suggesting that even larger basis results would increase the barrier corresponding to the high-symmetry linear configuration as compared to the bent configuration even further. This is the result regardless whether the MP2 or the CCSD(T) techniques are employed for the geometry optimizations. This means that for this particular complex the basis set providing meaningful results must be at least of aQZ quality and perhaps even larger.
Then comes the question whether the equilibrium structure of the T-shaped (-He)He(H
)He complex has a fully symmetric
or a less symmetric
equilibrium structure. For example, Balta and Gianturco [Citation57] made the following comment based on their electronic structure optimizations: ‘the (He–H–He)
interaction with an extra He atom presents a double well minimum potential with a T-shaped global minimum and a very shallow local minimum in the linear approach’. Our results clearly show that the equilibrium structure has
point-group symmetry: as expected, the He atom is attached to the formally linear He(H
)He core in a T-shaped form. The He–H–He angle decreases from
to
and the non-bonded He
H distance is 2.13 Å at the aug-cc-pVQZ CCSD(T) level. We have also discredited via higher-quality electronic-structure computations the existence of a linear local minimum found earlier by Balta and Gianturco [Citation57].
For the global nonlinear minimum the evaporation energy depends only very slightly on the extension of the basis. At the CCSD(T) level, for example, the electronic evaporation energies are 342.5, 351.0, and 354.1 cm−1 when the aug-cc-pVTZ, aug-cc-pVQZ, and the aug-cc-pV5Z basis sets are used, respectively, showing clear convergence to the CBS limit (again, as expected, the CCSD(T) – MP2 energy increments computed for the FPA analysis show extremely mild basis set dependence).
6.2. HHe
The naive expectation, without taking into account the attractive HeHe interaction, is that the equilibrium structure of the [0–2–0]-HHe
complex (see Figure ) has
point-group symmetry. Interestingly, this is indeed true when one employs the aug-cc-pVTZ basis at the MP2, CCSD, CCSD(T), and CCSDT levels. However, the
structure of [0–2–0]-HHe
is never a minimum when basis sets larger than aug-cc-pVTZ are utilised. With the aQZ and a5Z basis sets, the optimised
structures correspond to first-order transition states (TS), with a minuscule imaginary wavenumber of B
symmetry, between 5i
i cm−1 (the value gets larger as the basis gets larger). Motion along this mode makes the optimised equilibrium structure correspond to a very nearly
point-group symmetry structure.
With all basis sets used, the HHe distance of the core is extremely similar to that of the molecular cation He(H)He. For example, at the aug-cc-pVTZ MP2 level the equilibrium distance is shrinked to 0.92407 Å from 0.92409 Å. This means that the extra two He atoms attached to the core cause an extremely small perturbation to the electronic structure of the cationic core of the complex.
The simplicity of the structure of the [0–2–0]-HHe complex is reflected in its harmonic and anharmonic frequencies. There are the ‘usual’ four high-wavenumber modes above 800 cm−1, corresponding to the stretching and bending modes of the He(H
)He core (see Table ). The other five, bending modes of the complex are all below 200 cm−1.
6.3. HHe
The equilibrium structure of the [0–3–0]-HHe complex could have a maximum of
point-group symmetry. However, at all levels of electronic structure theory employed the equilibrium structure has lower,
point-group symmetry, as clearly seen in Figure . A similar asymmetry has been noted by de Lara-Castells and Mitrushchenkov [Citation81] for the CO
dopant in para-H
and by Whaley et al. in other cases [Citation122,Citation123], as well.
Figure 6. Equilibrium structure of the [0–3–0]-HHe complex, obtained at the aug-cc-pVTZ MP2 level, with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, denoted on the atoms (H is white, He is light blue).
![Figure 6. Equilibrium structure of the [0–3–0]-HHe5+ complex, obtained at the aug-cc-pVTZ MP2 level, with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, denoted on the atoms (H is white, He is light blue).](/cms/asset/1e9ef68c-c878-4753-8f04-5c40e71ccac0/tmph_a_1585984_f0006_oc.jpg)
The eight low-frequency modes of [0–3–0]-HHe are all below 200 cm−1, three of them are below 100 cm−1: 27 (
), 41 (
), and 89(
) cm−1 at the aug-cc-pVTZ CCSDT level. These are basically the same when computed at the aug-cc-pVTZ MP2 level (22, 35, and 85 cm−1, respectively). In fact, the values of all the fundamentals are stable across the different basis sets and electron-correlation levels. These results suggest that when for the larger complexes only the aug-cc-pVTZ MP2 level is used to obtain structural and spectroscopic results, they should be considered dependable.
6.4. HHe
The equilibrium structure of the [0–4–0]- complex has
point-group symmetry (see the left panel of Figure ); thus, it is a tetragonal bipyramid.
Figure 7. Structural isomers of HHe and HHe
, [0–4–0]-HHe
of
point-group symmetry (left), [0–5–0]-HHe
of
point-group symmetry (middle), and [1–4–0]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 7. Structural isomers of HHe6+ and HHe7+, [0–4–0]-HHe6+ of D4h point-group symmetry (left), [0–5–0]-HHe7+ of D5h point-group symmetry (middle), and [1–4–0]-HHe7+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/a0222d6b-bbfb-4103-9ef0-c21b18140065/tmph_a_1585984_f0007_oc.jpg)
Optimizations aimed at finding the [1–3–0]-HHe complex always ended up as [0–4–0]-HHe
, irrespective of the starting geometry and the level of electronic-structure theory applied. Thus, even if the [1–3–0]-HHe
isomer existed, certainly the barriers separating the two isomers must be small.
The [0–4–0]-HHe complex has four harmonic fundamentals below 100 cm−1, they are at 36 (
), 39 (
), and 66 (
) cm−1 at the aug-cc-pVTZ MP2 level. Again, there is a very clear separation of the core motions, with four harmonic fundamentals above 900 cm−1.
Our computed evaporation energies and ion trap measurements confirm one of the most important experimental MS results [Citation35,Citation39] concerning the HHe complexes, namely that the stability of the He-solvated complexes remains about the same for n=3−6 and decreases sharply after the HHe
complex. As Figure shows vividly, the evaporation energies for n=3−6 are basically the same, with or without the consideration of the ZPVE correction (though the correction itself is substantial, it decreases the evaporation energies from about 320 cm−1 to about 180 cm−1). If one compares Figure and , containing experimental and computed results, respectively basically the same gentle decrease in stability from n=3 to 6 can be observed. Thus, the computed evaporation energies support and explain the MS results of the present and previous studies.
6.5. HHe
The evaporation energy drops substantially for this complex but the equilibrium structure of the global minimum remains highly symmetric, of point-group symmetry, corresponding to a pentagonal bipyramid (middle panel of Figure ). This holds irrespective of which level of electronic-structure theory was employed for the optimisation of the structure.
In the [0–5–0]-HHe cation the buildup of the first solvation belt around the H
atom of the He–H
–He core becomes complete. The five-membered central belt is a recurring motif in the (global) minima of the larger He-solvated clusters.
The lowest-frequency, in fact doubly-degenerate (), vibrations characterising this structural motif are as large as 49, 48, and 48 cm−1 at the aTZ, aQZ, and a5Z MP2 levels, respectively. These values should be compared with the lowest-frequency values for [0–4–0]-HHe
, 36 and 26 cm−1 at the aug-cc-pVTZ and aug-cc-pVQZ MP2 levels, respectively. Nevertheless, adding a fifth He atom to the central ring does not yield the same stabilisation energy as building the central ring with up to 4 He atoms.
There is another HHe isomer, the [1–4–0]-HHe
cation, with an equilibrium structure of
point-group symmetry (see the right panel of Figure ). [1–4–0]-HHe
has a substantial relative electronic energy compared to the global minimum, 74.4 and 76.4 cm−1 at the aTZ and aQZ MP2 levels, respectively. Since the corresponding HF values are 27.9 and 29.6 cm−1, it is clear that consideration of electron correlation is extremely important to obtain correct relative energies. Once zero-point vibrations are taken into account, the energy difference between the two isomers drops from 74.4 to only 13.8 cm−1 at the aug-cc-pVTZ MP2 level. The similar relative energies and the extremely large-amplitude motions corresponding to the interconversion of the two isomers should make the internal dynamics of this complex an extremely interesting target for high-resolution molecular spectroscopy.
If one considers not the electronic but the ZPVE-corrected evaporation energies, which are systematically considerably lower than their electronic counterparts, the abrupt drop in the evaporation energy after HHe becomes pronounced (see Figure ). The ZPVE-corrected evaporation energies, about 180 cm−1 for up to n=6 drop to about 75 cm−1 between n=7−12. Then there is a further drop but this will be discussed later, when the structures and energetics of HHe
and HHe
are discussed.
6.6. HHe
There are several possible isomers for this complex with six solvating He atoms, as there are three distinct possibilities for the central belt: it could contain four, five, or six He atoms. The ‘planar’, point-group-symmetry [0–6–0] isomer has 3 imaginary modes at the aug-cc-pVTZ MP2 level. Isomers containing six He atoms in the central belt for
are thus deemed to be unfeasible and were not searched for in the larger complexes. The viable solvation shells for HHe
remain [1–5–0], [1–4–1], and [2–4–0], and these isomers of HHe
have indeed been found at the aug-cc-pVTZ MP2 level (see Figure ).
Figure 8. Structural isomers of HHe, [1–5–0]-HHe
(left, the global minimum), [2–4–0]-HHe
(middle), and [1–4–1]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 8. Structural isomers of HHe8+, [1–5–0]-HHe8+ (left, the global minimum), [2–4–0]-HHe8+ (middle), and [1–4–1]-HHe8+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/d3a96669-84a7-498c-955d-4c00093ad7a8/tmph_a_1585984_f0008_oc.jpg)
From the geometry optimizations it is clear that the five-atom central belt has the lowest energy, it is the global minimum at all levels of electronic-structure theory employed. Nevertheless, the two isomers with a 4-membered central belt have small relative energies.
[1–5–0]-HHe remains the global minimum-energy isomer irrespective whether one considers zero-point vibrations or not. However, the order of the [2–4–0] and [1–4–1] isomers is somewhat uncertain, as the difference in their electronic relative energies is only 5 cm−1. Due to the uncertainty in the computed (harmonic) ZPVE values, the correct energy order of the two isomers of HHe
with four He atoms in the central belt is not established definitively.
The internal dynamics of the HHe complex should be intriguing though it is unclear how the low-energy motions could be studied experimentally. There are four and nine harmonic fundamentals of [1–5–0]-HHe
below 50 and 100 cm−1, respectively, the lowest ones are at 13 and 24 cm−1. For [1–4–1]-HHe
, there are 10 harmonic fundamentals below 100 cm−1, as the He atoms of the two outer belts contribute two modes each.
As a result of the extra stability of the central belt of [1–5–0]-HHe compared to the 4-membered one, for the
complexes only the 5-atomic central belt has been considered. Occasionally, searches were performed to find 3-, 4-, and 6-membered central belts but without success.
6.7. HHe
Maintaining the five-fold central belt, there are two possible arrangements: [2–5–0]- and [1–5–1]-HHe (see Figure ). Both equilibrium structures seem to have
point-group symmetry. Note that [2–4–1]-HHe
also appears to be a minimum with
point-group symmetry but has a relative pure electronic energy higher by 77 cm−1 (at the aTZ MP2 level).
Figure 9. Structural isomers of HHe, [1–5–1]-HHe
(left), [2–5–0]-HHe
(middle), and [2–4–1]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 9. Structural isomers of HHe9+, [1–5–1]-HHe9+ (left), [2–5–0]-HHe9+ (middle), and [2–4–1]-HHe9+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/72f2514c-85d1-4c56-b9ef-7f5a064b6e31/tmph_a_1585984_f0009_oc.jpg)
While the solvating He atoms of the central belt have a partial Mulliken charge of only , those He atoms which solvate the core in the outer belts have an order of magnitude smaller charge (see Figure ). As always, the larger positive charge attracts the two extra solvating He atoms in the case of [2–5–0]-HHe
.
Which isomer has the lower energy depends on whether we consider the effect of zero-point vibrations or not. Without the ZPVE, [2–5–0]-HHe is the global minimum, though the stability difference is only 6.7 cm−1, while when ZPVE is considered, [1–5–1]–HHe
becomes lower in energy, but only by 1.7 cm−1, as the [1–5–1]–HHe
isomer has a harmonic ZPVE 8.4 cm−1 smaller than that of [2–5–0]-HHe
(Table ). The present computations are definitely not accurate enough to decide which isomer corresponds to the global minimum on the PES of HHe
.
6.8. HHe
There are two feasible structural isomers of HHe: [3–5–0] and [2–5–1]. Both isomers have been found at the aug-cc-pVTZ MP2 level (see Figure ).
Figure 10. Structural isomers of HHe, [3–5–0]-HHe
(left) and [2–5–1]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 10. Structural isomers of HHe10+, [3–5–0]-HHe10+ (left) and [2–5–1]-HHe10+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/8adbfa05-81a0-4a20-b618-3cd6a9931c05/tmph_a_1585984_f0010_oc.jpg)
Similar to HHe, the electronic and the zero-point-corrected order of the relative energies is different: consideration of ZPVE makes the [2–5–1] structure more stable by a substantial amount, 30.6 cm−1. Also similar to the case of HHe
, the isomer with the larger number of solvating He atoms in the outer belt has the larger ZPVE value, this time by a substantial 39.5 cm−1 (see Table ).
6.9. HHe
For HHe, the relative energies of the three structural isomers identified at the aug-cc-pVTZ MP2 level, [4–5–0]-, [3–5–1]-, and [2–5–2]-HHe
(see Figure ), change rather substantially depending on whether one considers ZPVE effects or not. This is due to the fact that the ZPVEs of the three isomers are significantly different, the difference between the two extreme cases, [4–5–0] and [2–5–2], is very large, 74.4 cm−1. As before, the more He atoms in the top solvation shell the larger the ZPVE. The pure electronic energy order of the isomers is [3–5–1], [2–5–2], and [4–5–0], with relative energies of 0.0, 3.3, and 38.6 cm−1, respectively (at the aug-cc-pVTZ MP2 level). After considering the ZPVEs of the isomers, the most ‘symmetric’ isomer, [2–5–2]-HHe
, becomes the global minimum.
Figure 11. Structural isomers of HHe, [4–5–0]-HHe
(left), [3–5–1]-HHe
(middle), and [2–5–2]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 11. Structural isomers of HHe11+, [4–5–0]-HHe11+ (left), [3–5–1]-HHe11+ (middle), and [2–5–2]-HHe11+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/cfaea785-ea09-4f75-bc72-a2f4209a04cf/tmph_a_1585984_f0011_oc.jpg)
At the first sight, the three identified isomers cannot interconvert easily into each other. Nevertheless, all of the isomers have a large number of low-frequency harmonic modes. Some of the associated motions should facilitate the facile interconversion of the isomers in the [4–5–0]–[3–5–1]–[2–5–2] sequence, through appropriate large-amplitude motions. Spectroscopic characterisation of the nuclear dynamics would be extremely interesting.
6.10. HHe
Two HHe isomers with a central belt of five He atoms have been found at the aug-cc-pVTZ MP2 level: [4–5–1]- and [3–5–2]-HHe
(see Figure ). A third possible isomer, [5–5–0]-HHe
, could not be located at the aug-cc-pVTZ MP2 level. This is taken as an indication that while the central belt preferentially contains five solvating He atoms, the outer belts, ‘supported’ by considerably smaller positive partial charges, may have a maximum of four He atoms (plus several capping possibilities to accomodate more He atoms).
Figure 12. Two structural isomers of the HHe complex, [4–5–1]-HHe
(left) and [3–5–2]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 12. Two structural isomers of the HHe12+ complex, [4–5–1]-HHe12+ (left) and [3–5–2]-HHe12+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/239d608a-57d9-4619-ba66-c0eb7ac7c2e0/tmph_a_1585984_f0012_oc.jpg)
There is again a substantial difference in the ZPVE estimates of the isomers, and once again the isomer with the larger number of solvating He atoms in the top shell has the larger ZPVE, this time by 41.7 cm−1 (Table ). Nevertheless, consideration of the ZPVE corrections does not change the relative-energy order of the isomers.
6.11. HHe
The three structural isomers of HHe found, at the aug-cc-pVTZ MP2 level, during this computational investigation are depicted in Figure . This is the first time that a capping He atom appears in one of the outer belts (see the right panel of Figure ). The global minimum is the [3–5–3]-HHe
complex.
Figure 13. Three structural isomers of the HHe complex, [3–5–3]-HHe
(left), [4–5–2]-HHe
(middle), and [14–5–1]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 13. Three structural isomers of the HHe13+ complex, [3–5–3]-HHe13+ (left), [4–5–2]-HHe13+ (middle), and [14–5–1]-HHe13+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/aa9901ca-353a-4d65-bd22-3321cb8155bc/tmph_a_1585984_f0013_oc.jpg)
According to high-quality, high-resolution experimental MS studies [Citation35,Citation39], this is the complex after which the evaporation energies should drop substantially once again (the previous drop was after n=6, discussed above). Nevertheless, this is not quite the picture which emerges from our computations. Starting with HHe, the pure electronic evaporation energy is about 140 cm−1 at the aug-cc-pVTZ MP2 level (should be higher for more extended electron-correlation treatments and much lower, see Figure , if ZPVE is considered). Note that for HHe
the electronic evaporation energy is 328 cm−1, while for HHe
it is 216 cm−1. The purely electronic evaporation energies remain close to 140 cm−1 for the global minima between n=8−13 (at the aug-cc-pVTZ MP2 level). If the ZPVE effect is considered, one may say that the drop in the evaporation energies happens after n=13, as observed experimentally. Note, furthermore, that it is the n=13 complex where the Hartree–Fock (HF) evaporation energy is substantially higher than zero, for
the complexes do not seem to be bound at the HF level.
One may consider the capping of the outer belt in [14–5–1]-HHe as the starting point of the buildup of even another solvation shell, beyond the three principal belts. The He atom farthest away from the central proton indeed has a Mulliken charge of 0.000, suggesting its extremely loose attachment to the upper belt, whose He atoms possess partial Mulliken charges of about
.
6.12. HHe
Two minima have been identified for this cluster at the aug-cc-pVTZ MP2 level: [4–5–3] is the global minimum and [4–5–2] is a local minimum (see Figure ).
Figure 14. Two structural isomers of the HHe complex, [4–5–3]-HHe
(left) and [4
–5–2]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 14. Two structural isomers of the HHe14+ complex, [4–5–3]-HHe14+ (left) and [41–5–2]-HHe14+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/a5628004-ba96-4f5e-9130-84975bf813e3/tmph_a_1585984_f0014_oc.jpg)
The global minimum still has a substantial electronic evaporation energy at the MP2 level but it is significantly smaller than the n=13 value.
6.13. HHe
There are two structural isomers identified for this complex at the aug-cc-pVTZ MP2 level: [4–5–3]- and [4–5–4]-HHe
(see Figure ). Thus, this is the second time when an outer belt is solvated in an ‘in-plane’ fashion resulting in an isomer.
Figure 15. Two structural isomers of the HHe complex, [4
–5–3]-HHe
(left) and [4–5–4]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 15. Two structural isomers of the HHe15+ complex, [41–5–3]-HHe15+ (left) and [4–5–4]-HHe15+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/3f80a970-83b5-43db-8389-08d13db0f750/tmph_a_1585984_f0015_oc.jpg)
It is not straightforward to distinguish a ‘’ motif from a ‘5’ one, but the partial Mulliken charges help doing this. As a guide, we can safely assume that the partial charges within a belt are similar, though they may differ strongly from those of another belt. For example, in the HHe
case the central belt has Mulliken charges of about
, while the outer belts have smaller partial charges of about
. Then, in the case of [4
–5–3]-HHe
we see a partial charge of about 0.000 for the capping He. Clearly, this He is not directly part of a belt governed by a He
core atom but it solvates two of the He atoms of the belt in an
fashion.
This is seemingly the first case when the HF evaporation energy, computed at the aug-cc-pVTZ MP2 optimised structure not only tends to be close to zero but becomes negative. This happens for the [4–5–4] complex (see Figure ).
Based on previous structural observations about the inner and outer belts, [4–5–4]-HHe is expected to be the complex where the three belts corresponding to the He
–H
–He
core are maximally filled up. Somewhat surprisingly, this structural singularity is not reflected by the relative energies.
6.14. HHe
This is an interesting cluster from the point of view that it exhibits, for the first time, a [4–5–4]-like belt system with two different types of solvated 4-membered outer rings (see Figure ). No other types of isomers could be located on the PES of HHe, not at least at the aug-cc-pVTZ MP2 level.
Figure 16. Two structural isomers of the HHe complex, [4
–5–4]-HHe
(left) and [14–5–4]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 16. Two structural isomers of the HHe16+ complex, [41–5–4]-HHe16+ (left) and [14–5–4]-HHe16+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/c53431af-ecfa-4e51-af49-203409e78bb5/tmph_a_1585984_f0016_oc.jpg)
In the global minimum, a solvating He is attached to the 4-membered ring in a ‘co-planar’ fashion, while in the secondary minimum the solvating He atom caps one of the 4-membered outer He belts, situated around the He atom of the core having the larger positive charge. Thus, the global and the secondary minima exhibit [–5–4] and [14–5–4] solvation shells, respectively. As emphasized before, since one of the He atoms in the upper belt has a partial charge of 0.000 and the others have about
, it is clearly better to call this motif ‘
’ rather than ‘5’.
As expected, the energy difference between the two isomers is not substantial, only 11.7 cm−1 at the aug-cc-pVTZ MP2 level without the ZPVE correction. Consideration of the ZPVE corrections does not change the ordering of the two isomers.
6.15. HHe
Two structural isomers have been identified for HHe, [14–5–
] and [14–5–14] (see Figure ). At the HF level the evaporation energy is negative but at the MP2 level the evaporation energy is still substantial, 50 cm−1 without consideration of the ZPVEs. The [
–5–41]-HHe
isomer appears to be the global minimum.
Figure 17. Two structural isomers of the HHe complex, [14–5–14]-HHe
(left) and [14–5–
]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 17. Two structural isomers of the HHe17+ complex, [14–5–14]-HHe17+ (left) and [14–5–41]-HHe17+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/a0ed69e1-dc9e-4706-b517-9fc5143672d2/tmph_a_1585984_f0017_oc.jpg)
6.16. HHe
There are three similar structural isomers we were able to identify for this cluster at the aug-cc-pVTZ MP2 level (see Figure ). These isomers exhibit a five-membered middle belt around H, and one 14- and one
-capped outer belt.
Figure 18. Three structural isomers of the HHe complex, [
–5–14]-HHe
(left), [
–5–14]-HHe
(middle), and [
–5–14]-HHe
(right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).
![Figure 18. Three structural isomers of the HHe18+ complex, [41,1,s–5–14]-HHe18+ (left), [41,1,a–5–14]-HHe18+ (middle), and [141–5–14]-HHe18+ (right), with Mulliken charges, obtained at the aug-cc-pVTZ RHF level, given on the atoms (H is white, He is light blue).](/cms/asset/92f94e45-6c48-48af-b7fd-47b98311658d/tmph_a_1585984_f0018_oc.jpg)
The global minimum is [–5–14]-HHe
. The upper ring actually looks quite like a basin, the Mulliken charges help to identify the bonding motifs once again. The second most stable structure, after considering ZPVE effects, contains a 4-membered ring with two ‘co-planar’ solvating He on opposite sides. This complex can be denoted as
following the notation introduced in Section 5. The least stable structure has a 4-membered ring with both a capping and a ‘co-planar’ He, denoted accordingly as
. The minuscule energy differences between the three minima suggest a very complex internal dynamics characterising HHe
.
7. Signatures of microscopic superfluidity
In this section we go through possible signatures of microscopic superfluidity which the electronic-structure computations of this study reveal. The MS measurements of this study, while useful for other purposes, are not able to say anything about MSF, as they stop at n=7. Thus, only some of the computational results are considered below.
7.1. Evaporation energies
Table contains the evaporation energies of the HHe species considered in this study, obtained at different levels of electronic-structure theory (HF, MP2, and CCSD(T)) using the aug-cc-pVTZ, aug-cc-pVQZ, and aug-cc-pV5Z basis sets.
The evaporation energies of HHe, HHe
, and HHe
, about 320 cm−1 at the aug-cc-pVTZ MP2 level and without the ZPVE (see Table ), are almost the same as that of He(H
)He·He. This could be understood simply by the fact that in these cases building up of the first solvation belt, that around the central proton, is taking place. Since the relative ZPVE contributions of (
He
)He(H
)He,
, are about the same at about 130 cm−1 (see Table ), all these cations have a
of about 190 cm−1. As the vibrational excitations have a very small energy (e.g. the three extra vibrational modes per added He atom make up a 140 cm−1 increment), there will be a large number of bound rovibrational states for all these species. This means a rich rovibrational dynamics worth studying by techniques of high-resolution molecular spectroscopy. The electronic evaporation energy drops significantly for n=7, but it is still substantial. It is for the n=7 complex that filling of the inner belt is completed. These cations,
, obviously show no signs of MSF, these complexes appear to be ‘molecule like’. As to the nuclear dynamics of HHe
, it is enriched by the presence of two isomers (complexes n=3−6 seemingly possess only a single minimum on their PESs).
The next set of similar electronic evaporation energies, around 140 cm−1, is between n=8 and 13. Note that if ZPVE is considered, n=7 also belongs to this set, where filling of the outer solvation belts take place. This is a somewhat surprising result and suggests that in HHe complex stability and solvation-shell structure do not go hand in hand. These complexes, all possessing at least two isomers, are not expected to show signs of MSF.
Then, after n=13, the HF evaporation energies become minuscule, suggesting that most of the binding is due to electron correlation. This is the place where the electrostatic interaction between the substantial positive partial charges of the atoms of the quasilinear core with the hardly polarisable He atoms play less and less role in binding further He atoms. In other words, this is the point where one can start considering looking for signs of MSF. At we can observe the buildup of further ‘solvation shells’, with 14 and
structural motifs. The ZPVE-corrected evaporation energies also approach the bulk value of a few cm−1 here. This suggests that one can search for experimental signs of MSF starting from complexes having
.
7.2. Structural parameters and rotational constants
It cannot be expected that explicit structural parameters prove to be useful indicators of MSF. Nevertheless, rotational constants, directly related to structures of molecules, have been used to judge the onset of MSF. Thus, it is useful to collect the rotational constants of the HHe species, especially for
, to see if they show any trends. The rotational constants
,
, and
of the HHe
complexes, obtained at the aug-cc-pVTZ MP2 level and corresponding to the rigid-rotor model, are collected in Table .
The value of the linear HHe
chromophore can serve as a reference to the rotational constant values of the larger complexes. Clearly, up to n=18 the rotational constants become smaller and smaller as the moments of inertia become larger with solvating He atoms farther and farther away from the proton of the complex.
Deviations of the experimental effective rotational constants, used to simulate observed experimental spectra, from the present rigid-rotor values could be useful signatures of MSF (vide infra). Most of the complexes are near symmetric tops, pointing toward a somewhat simpler possible interpretation of future high-resolution spectroscopic measurements. The feasibility of such MW experiments applying novel ion-trap-based double-resonance schemes [Citation92,Citation93] has been sketched in Section 2.
As discussed before, rotational constants for rigid molecular structures decrease with n and deviations from the rigid-rotor picture could be taken as indicators of MSF. For all the HHe ions, the predicted structures contain the HHe
chromophore, and when this chromophore is surrounded by more and more He atoms clear progressions for the rotational constants are expected and observed. It is an open question to what extent large-amplitude motions of the surrounding He atoms will change the effective rotational constants. High-resolution spectroscopic investigation of these clusters is thus desirable and it also seems feasible, as our preliminary experimental work (not detailed here) indicates.
For the larger clusters more spectral features will be arising because the structures depart from the prolate symmetric-top limit and thus an experimental asymmetry in the rotational constants, defined as B−C, may provide insight into the true molecular structure and about the influence of large-amplitude motions. A very small asymmetry has been found in the rotational spectra of the He-CH complex [Citation92], even though the large-amplitude motion of He with respect to the CH
molecule should lead to the spectrum of a prolate symmetric-top molecule. This result suggests that spectroscopic surprises are expected already for the smaller HHe
complexes.
Based on the quantum-chemical computation of the structures of the larger complexes, n>6, internal rotations are expected to complicate the observed spectra. Depending on the energy landscape of the HHe chromophore versus the surrounding He atoms also free internal rotations could be present, which would complicate the rotational structure of the spectral bands. On the other hand, this might give insight into the large-amplitude motions and their influence on the effective rotational constants, as discussed above. Interesting insight into the interplay between the internal and the end-over-end rotations could also be gained. In the end such a detailed picture could help to distinguish between complex internal motions and the onset of MSF in the HHe
clusters.
7.3. Vibrations
It is expected that as n increases the movements of the solvating He atoms become more and more unconstrained by the core. As a result, the harmonic frequencies of the normal modes would become lower and lower. It is not expected that the harmonic frequencies represent well the extremely anharmonic motions of the cations with larger n values but they may give some indication about the onset of MSF, especially since we are lacking better indicators as long as no detailed dynamical studies are performed.
In all complexes investigated the internal motions of the quasilinear triatomic core have four high frequencies, above 900 cm−1 in all but one case (894 cm−1 for HHe), separated well from all the other 3N−10 modes. The changes in these frequencies by changing n are small (see Table ) and it may not seem feasible to learn much about microscopic superfluidity from these harmonic (or anharmonic) frequencies. The best one can hope is that if basically no changes are observed for them it means that the movement of an additional He atom can be envisioned as being free, sort of a requirement toward MSF. This seems to be the case at about n=16, where the basic triple-belt structure [4–5–4] of the HHe
complexes is completed and the additional solvating He atoms form caps or
-type bonds to the He atoms of the outer belts.
8. Summary and conclusions
The equilibrium structures and the vibrational dynamics of HHe species have been investigated for n=1−18 via high-level electronic-structure computations. Cryogenic ion-trap experiments have also been performed as part of this study, to supplement the computations; HHe
ions were observed up to n=7 but not beyond.
The special stability of HHe is shown experimentally in this work for the first time (see Figure ), while that of n=6 [Citation35,Citation39] is reproduced by our experiments. The first spectroscopic experiments we performed at the FELIX facility [Citation91] indeed confirm the role the triatomic quasilinear He(H
)He unit plays as the chromophore for larger HHe
complexes. This is also clearly established by our computational results, which yield for the chromophore substantial partial charges of about +0.56 on H and about +0.22 on the two core He atoms. Electrostatic interaction between these large positive charges and the hardly polarisable solvating He atoms are responsible for the structures of the smaller members of the HHe
family. This is also reflected by the fact that solvating He atoms often acquire a small negative charge and more He atoms are attached to the He of the core carrying the larger positive partial charge.
The strongly-bound complexes HHe and HHe
could be studied with the highest levels of electronic structure theory (up to FCI), including focal-point analyses (FPA) of some relative energies, yielding appropriate uncertainty estimates for the computed energy differences. The definitive quantum-chemical results obtained include the proton affinity of He, computed to be
cm−1, the isomerisation energy between the two linear forms of HHe
,
cm−1, and the dissociation energy of the HHe
HHe
+ He reaction, with an FPA estimate of
cm−1.
Since the structure of all HHe clusters can be described by three main solvation belts around the three atoms of the HHe
chromophore, a useful description of the structures of the complexes is provided by the [k−l−m]-HHe
notation, where l denotes the number of solvating He atoms in the central belt and
denote the number of solvating He atoms in the outer (top and bottom) belts. He atoms capping the top and bottom belts, arising first for the n=13 case, can be indicated by adding sub- and superscripts to k and m when extra He atoms cap the ring or attach to two atoms of the ring, respectively.
For the n=3−5 cations the equilibrium structures exhibit lowering of the symmetry from the ideal ones. For example, the equilibrium structure of the T-shaped molecular cation HHe and that of HHe
have
point-group symmetry. Due to the weak attractive He
He interaction, the n=3−5 cations appear to be polytopic. Determination of their correct equilibrium structure requires highly correlated techniques of electronic structure theory, extended basis sets, and a careful checking of the computed results.
We confirm the experimental MS results about the extra relative stability of complexes up to n=6. In all the n=3−6 complexes the central belt around the proton is building up. The equilibrium structure of the HHe complex has
point-group symmetry. Interestingly, building up of the central belt is continued for n=7, where the equilibrium structure has
point-group symmetry. Nevertheless, the evaporation energy of HHe
is considerably smaller than that of HHe
. This means that the larger stability of the complexes up to n=6 is not simply due to the structural feature of the building of the central belt. The 5-membered central belt motif will occur in almost all of the isomers of the clusters having larger n values.
The reason of the extra stability of the n=13 complex compared to the n=14 one is somewhat less clear. By this time the outer belts are formed, the global minimum of the HHe cluster can be characterised as [3–5–3]-HHe
. Thus, we are two helium atoms away from the filling of the three belts, i.e. from the [4–5–4] configuration. Seemingly in this case the stability change is once again not related directly to the filling of the shell structure of the HHe
series.
While for the strongly bound molecular cations, HHe and HHe
, where the structure is determined mostly by electrostatic interaction, HF theory provides about 90% of the binding energy, for the He-solvated species a large part of the evaporation energy originates from electron correlation. For smaller n values, about 50% of the evaporation energy is coming from the MP2 increment. Starting from the buildup of the second solvation belt (n=8), forming around the ‘more positive’ He atom of the core, HF theory struggles to yield even semiquantitative evaporation energies. From around n=13, HF theory is unable to say anything useful about the stability of the complexes; in these cases it is the electron correlation that binds the complexes.
It is of interest to review the quantum effect of ZPVE on the stability of He-solvated complexes. First, the ZPVE correction is substantial, can be 2/3 of the pure electronic evaporation energy. Since in the He-solvated proton complexes the harmonic picture may not be a very good approximation and second-order vibrational perturbation theory may also be unreliable for modes with large-amplitude motions, variational computation of the ZPVE is desirable. This may not be simple by some of the direct variational techniques [Citation120] and generating accurate PESs for these systems is also far from being trivial due partly to the polytopic nature of these species. Second, while the ZPVE correction may be as large as the electronic evaporation energy (see Figure ), all of the investigated species do exist under suitable circumstances. This is only true when electron correlation is taken into account, adding further electron correlation to the treatment yields larger electronic evaporation energies. Thus, it is expected that these complexes with n values considerably larger than 18 do exist. This is in accord with the results of high-resolution MS studies [Citation39] which could see HHe complexes up to n=30. Third, ZPVE-corrected evaporation energies are somewhat more similar than their electronic counterparts during the buildup of the belts. In fact, as Figure demonstrates, there is a substantial drop at the otherwise relatively steady evaporation energies at n=6 and n=13, supporting related MS and cryogenic ion trap experimental results, and after about n=13 the ZPVE-corrected evaporation energies tend to be close to zero, suggesting an eventual onset of microscopic superfluidity.
Note, finally, that the present computational and cryogenic ion trap experimental investigation could be extended to other cationic clusters containing hydrogen and helium. These clusters would involve H or H
in the core. H
He
has been the subject of some advanced theoretical studies [Citation68,Citation69,Citation124], while H
He
has already been synthesised and investigated spectroscopically in the same trap machine as used in this study [Citation67]. Further quantum chemical and low- as well as high-resolution spectroscopic studies on these species are planned in our laboratories.
Acknowledgments
Discussions with Dr. Maria Pilar de Lara-Castells on the topic of microscopic superfluidity helped to form this paper. Advice of Prof. Mihály Kállay on convergence criteria concerning the CFOUR and MRCC electronic-structure packages is gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Attila G. Császár http://orcid.org/0000-0001-5640-191X
Tamás Szidarovszky http://orcid.org/0000-0003-0878-5212
Oskar Asvany http://orcid.org/0000-0003-2995-0803
Stephan Schlemmer http://orcid.org/0000-0002-1421-7281
Additional information
Funding
Related Research Data
References
- S. Goyal, D.L. Schutt and G. Scoles, Phys. Rev. Lett. 69, 933 (1992). doi: 10.1103/PhysRevLett.69.933
- S. Goyal, D.L. Schutt and G. Scoles, Phys. Rev. Lett. 73, 2512 (1994). doi: 10.1103/PhysRevLett.73.2512
- M.Y. Choi, G.E. Douberly, T.M. Falconer, W.K. Lewis, C.M. Lindsay, J.M. Merritt, P.L. Stiles and R.E. Miller, Int. Rev. Phys. Chem. 25, 15–75 (2006). doi: 10.1080/01442350600625092
- J.P. Toennies, Mol. Phys. 111, 1879–1891 (2013). doi: 10.1080/00268976.2013.802039
- K.K. Lehmann and G. Scoles, Science 279, 2065–2066 (1998). doi: 10.1126/science.279.5359.2065
- P. Sindzingre, M.L. Klein and D.M. Ceperley, Phys. Rev. Lett. 63, 1601–1604 (1989). doi: 10.1103/PhysRevLett.63.1601
- S.A. Chin and E. Krotscheck, Phys. Rev. B 52, 10405 (1995). doi: 10.1103/PhysRevB.52.10405
- S. Grebenev, J.P. Toennies and A.F. Vilesov, Science 279, 2082–2086 (1998). doi: 10.1126/science.279.5359.2083
- Y. Kwon, P. Huang, M.V. Patel, D. Blume and K.B. Whaley, J. Chem. Phys. 113, 6469 (2000). doi: 10.1063/1.1310608
- A. Stace, Science 294, 1292–1293 (2001). doi: 10.1126/science.1066731
- J. Tang, Y. Xu, A.R.W. McKellar and W. Jäger, Science 297, 2030–2033 (2002). doi: 10.1126/science.1073718
- J. Jortner, J. Chem. Phys. 119, 11335 (2003). doi: 10.1063/1.1622651
- Y. Xu and W. Jäger, J. Chem. Phys. 119, 5457–5466 (2003). doi: 10.1063/1.1598953
- S. Moroni, A. Sarsa, S. Fantoni, K.E. Schmidt and S. Baroni, Phys. Rev. Lett. 90, 143401 (2003). doi: 10.1103/PhysRevLett.90.143401
- F. Paesani, A. Viel, F.A. Gianturco and K.B. Whaley, Phys. Rev. Lett. 90, 073401 (2004).
- J.P. Toennies and A.F. Vilesov, Angew. Chem. Int. Ed. 43, 2622–2648 (2004). doi: 10.1002/anie.200300611
- S. Moroni, N. Blinov and P.-N. Roy, J. Chem. Phys. 121, 3577–3581 (2004). doi: 10.1063/1.1774160
- W. Topic, W. Jäger, N. Blinov, P.-N. Roy, M. Botti and S. Moroni, J. Chem. Phys. 125, 144310 (2006). doi: 10.1063/1.2357604
- S. Grebenev, B. Sartakov, J.P. Toennies and A.F. Vilesov, Science 289, 1532–1535 (2000). doi: 10.1126/science.289.5484.1532
- E.W. Draeger and D.M. Ceperley, Phys. Rev. Lett. 90, 065301 (2003).
- P. Sindzingre, D.M. Ceperley and M.L. Klein, Phys. Rev. Lett. 67, 1871–1874 (1991). doi: 10.1103/PhysRevLett.67.1871
- H. Li, R.J. Le Roy, P.-N. Roy and A. McKellar, Phys. Rev. Lett. 105, 133401 (2010).
- B.E. Callicoatt, D.D. Mar, V.A. Apkarian and K.C. Janda, J. Chem. Phys. 105, 7872 (1996). doi: 10.1063/1.472567
- M. Fárník, U. Henne, B. Samelin and J.P. Toennies, Z. Phys. D 40, 93–98 (1997). doi: 10.1007/s004600050166
- M. Fárník, U. Henne, B. Samelin and J.P. Toennies, Phys. Rev. Lett. 81, 3892 (1998). doi: 10.1103/PhysRevLett.81.3892
- B.E. Callicoatt, K. Förde, T. Ruchti, L.L. Jung, K.C. Janda and N. Halberstadt, J. Chem. Phys.108, 9371 (1998). doi: 10.1063/1.476389
- N. Halberstadt and K.C. Janda, Chem. Phys. Lett. 282, 409–412 (1998). doi: 10.1016/S0009-2614(97)01276-1
- J. Seong, K.C. Janda, N. Halberstadt and F. Spiegelmann, J. Chem. Phys. 109, 10873 (1998). doi: 10.1063/1.477784
- T. Ruchti, K. Förde, B.E. Callicoatt, H. Ludwigs and K.C. Janda, J. Chem. Phys. 109, 10679 (1998). doi: 10.1063/1.477765
- U. Henne and J.P. Toennies, J. Chem. Phys. 108, 9327 (1998). doi: 10.1063/1.476385
- M. Rastogi, C. Leidlmair, L. An der Lan, J. Ortiz de Zárate, R. Pérez de Tudela, M. Bartolomei, M.I. Hernández, J. Campos-Martínez, T. González-Lezana, J. Hernández-Rojas, J. Bretón, P. Scheier and M. Gatchell, Phys. Chem. Chem. Phys. 20, 25569–25576 (2018). doi: 10.1039/C8CP04522D
- D. Gerlich, J. Chin. Chem. Soc. 65, 637–653 (2018). doi: 10.1002/jccs.201800122
- T.R. Hogness and E.G. Lunn, Phys. Rev. 26, 44 (1925). doi: 10.1103/PhysRev.26.44
- G.E. Veatch and H.J. Oskam, Phys. Lett. A 28, 740–741 (1969). doi: 10.1016/0375-9601(69)90594-5
- T.M. Kojima, N. Kobayashi and Y. Kaneko, Z. Phys. D 23, 181–185 (1992). doi: 10.1007/BF01436742
- F. Grandinetto, Int. J. Mass. Spectrom. 237, 243–267 (2004). doi: 10.1016/j.ijms.2004.07.012
- S. Jaksch, A. Mauracher, A. Bacher, S. Denifl, F. Ferreira da Silva, H. Schöbel, O. Echt, T.D. Märk, M. Probst, D.K. Bohme and P. Scheier, J. Chem. Phys. 129, 224306 (2008). doi: 10.1063/1.3035833
- S. Jaksch, F.F. da Silva, S. Denifl, O. Echt, T.D. Märk and P. Scheier, Chem. Eur. J. 15, 4190–4194 (2009). doi: 10.1002/chem.200802545
- P. Bartl, C. Leidlmair, S. Denifl, P. Scheier and O. Echt, Chem. Phys. Chem. 14, 227–232 (2013). doi: 10.1002/cphc.201200664
- H.H. Michels, J. Chem. Phys. 44, 3834 (1966). doi: 10.1063/1.1726541
- D.E. Tolliver, G.A. Kyrala and W.H. Wing, Phys. Rev. Lett. 43, 1719 (1979). doi: 10.1103/PhysRevLett.43.1719
- D.M. Bishop and L.M. Cheung, J. Mol. Spectrosc. 75, 462–473 (1979). doi: 10.1016/0022-2852(79)90090-0
- A. Carrington, J. Buttenshaw, R.A. Kennedy and T.P. Softley, Mol. Phys. 44, 1233–1237 (1981). doi: 10.1080/00268978100103151
- P. Bernath and T. Amano, Phys. Rev. Lett. 48, 20 (1982). doi: 10.1103/PhysRevLett.48.20
- A. Carrington, R.A. Kennedy, T.P. Softley, P.G. Fournier and E.G. Richard, Chem. Phys. 81, 251–261 (1983). doi: 10.1016/0301-0104(83)85319-1
- D.-J. Liu, W.-C. Ho and T. Oka, J. Chem. Phys. 87, 2442 (1987). doi: 10.1063/1.453084
- J. Tennyson and S. Miller, J. Chem. Phys. 87, 6648–6652 (1987). doi: 10.1063/1.453399
- M.W. Crofton, R.S. Altman, N.N. Haese and T. Oka, J. Chem. Phys. 91, 5882 (1989). doi: 10.1063/1.457456
- J. Purder, S. Civiš, C.E. Blom and M.C. van Hemert, J. Mol. Spectrosc. 153, 701–709 (1992). doi: 10.1016/0022-2852(92)90504-H
- A. Carrington, D.I. Gammie, A.M. Shaw, S.M. Taylor and J.M. Hutson, Chem. Phys. Lett. 260, 395–405 (1996). doi: 10.1016/0009-2614(96)00860-3
- M. Juřek, V. Špirko, W.P. Kraemer, J. Mol. Spectrosc. 182, 364–370 (1997). doi: 10.1006/jmsp.1996.7236
- Z. Liu and P.B. Davies, J. Chem. Phys. 107, 337 (1997). doi: 10.1063/1.474394
- F. Matsushima, T. Oka and K. Takagi, Phys. Rev. Lett. 78, 1664–1666 (1997). doi: 10.1103/PhysRevLett.78.1664
- F. Filippone and F.A. Gianturco, Europhys. Lett. 44, 585 (1998). doi: 10.1209/epl/i1998-00513-x
- F.A. Gianturco and F. Filippone, Chem. Phys. 241, 203–212 (1999). doi: 10.1016/S0301-0104(98)00388-7
- I. Baccarelli, F.A. Gianturco and F. Schneider, Int. J. Quant. Chem. 74, 193 (1999). doi: 10.1002/(SICI)1097-461X(1999)74:2<193::AID-QUA15>3.0.CO;2-O
- B. Balta and F.A. Gianturco, Chem. Phys. 254, 203–213 (2000). doi: 10.1016/S0301-0104(00)00037-9
- B. Balta, F.A. Gianturco and F. Paesani, Chem. Phys. 254, 215–229 (2000). doi: 10.1016/S0301-0104(00)00038-0
- W.P. Kraemer, V. Špirko and O. Bludský, Chem. Phys. 276, 225–242 (2002). doi: 10.1016/S0301-0104(01)00694-2
- M. Šindelka, V. Špirko and W.P. Kraemer, Theor. Chem. Acc. 110, 170–175 (2003). doi: 10.1007/s00214-003-0468-9
- M. Pavanello, S. Bubin, M. Molski and L. Adamowicz, J. Chem. Phys. 123, 104306 (2005). doi: 10.1063/1.2012332
- M. Stanke, D. Kedziera, M. Molski, S. Bubin, M. Barysz and L. Adamowicz, Phys. Rev. Lett. 96, 233002 (2006). doi: 10.1103/PhysRevLett.96.233002
- A. Chakraborty, S. Giri and P.K. Chattaraj, New J. Chem. 34, 1936–1945 (2010). doi: 10.1039/c0nj00040j
- D. De Fazio, M. de Castro-Vitores, A. Aguado, V. Aquilanti and S. Cavalli, J. Chem. Phys. 137, 244306 (2012). doi: 10.1063/1.4772651
- W. Tung, M. Pavanello and L. Adamowicz, J. Chem. Phys. 137, 164305 (2012). doi: 10.1063/1.4759077
- A.J. Perry, J.N. Hodges, C.R. Markus, G.S. Kocheril and B.J. McCall, J. Chem. Phys. 141, 101101 (2014). doi: 10.1063/1.4895505
- I. Savić, D. Gerlich, O. Asvany, P. Jusko and S. Schlemmer, Mol. Phys. 113, 2320–2332 (2015). doi: 10.1080/00268976.2015.1037802
- T. Szidarovszky and K. Yamanouchi, Mol. Phys. 115, 1916–1926 (2017). doi: 10.1080/00268976.2017.1297863
- D. Papp, T. Szidarovszky and A.G. Császár, J. Chem. Phys. 147, 094106 (2017). doi: 10.1063/1.5000680
- M. Beyer, A. Lammers, E.V. Savchenko, G. Niedner-Schatteburg and V.E. Bondybey, Phys. Chem. Chem. Phys. 1, 2213–2221 (1999). doi: 10.1039/a809480b
- T.H. Dunning Jr., J. Chem. Phys. 90, 1007–1023 (1989). doi: 10.1063/1.456153
- T. van Mourik, A.K. Wilson and T.H. Dunning Jr., Mol. Phys. 99, 529–547 (1999). doi: 10.1080/00268979909482990
- F.A. Gianturco and F. Filippone, Comp. Phys. Comm. 145, 78–96 (2002). doi: 10.1016/S0010-4655(02)00148-0
- W.-D. Stohrer and R. Hoffmann, J. Am. Chem. Soc. 94, 1661–1668 (1972). doi: 10.1021/ja00760a039
- E. Clementi, H. Kistenmacher and H. Popkie, J. Chem. Phys. 58, 2460–2466 (1973). doi: 10.1063/1.1679525
- I.M.B. Nielsen, W.D. Allen, A.G. Császár and H.F. Schaefer, J. Chem. Phys. 107, 1195–1211 (1997). doi: 10.1063/1.474612
- P. Jungwirth and A. Krylov, J. Chem. Phys. 115, 10214–10219 (2001). doi: 10.1063/1.1409355
- M.P. de Lara-Castells, D. López-Durán, G. Delgado-Barrio, P. Villarreal, C. Di Paola, F.A. Gianturco and J. Jellinek, Phys. Rev. 71, 033203 (2005). doi: 10.1103/PhysRevA.71.033203
- M.I. Hernández, N. Halberstadt, W.D. Sands and K.C. Janda, J. Chem. Phys. 113, 7252–7267 (2000). doi: 10.1063/1.1313786
- P.M. Felker, J. Chem. Phys. 125, 184313 (2006). doi: 10.1063/1.2371032
- M.P. de Lara-Castells and A.O. Mitrushchenkov, J. Phys. Chem. Lett. 2, 2145–2151 (2011). doi: 10.1021/jz200801r
- O. Asvany, F. Bielau, D. Moratschke, J. Krause and S. Schlemmer, Rev. Sci. Instr. 81, 076102 (2010). doi: 10.1063/1.3460265
- O. Asvany, S. Brünken, L. Kluge and S. Schlemmer, Appl. Phys. B 114, 203–211 (2014). doi: 10.1007/s00340-013-5684-y
- W.A. Chupka and M.E. Russell, J. Chem. Phys. 49, 5426–5437 (1968). doi: 10.1063/1.1670068
- O. Asvany, K.M.T. Yamada, S. Brünken, A. Potapov and S. Schlemmer, Science 347, 1346–1349 (2015). doi: 10.1126/science.aaa3304
- H. Kohguchi, K.M.T. Yamada, P. Jusko, S. Schlemmer and O. Asvany, J. Chem. Phys. 148, 144303 (2018). doi: 10.1063/1.5023633
- J.L. Doménech, P. Jusko, S. Schlemmer and O. Asvany, Astrophys. J. 857, 61 (2018). doi: 10.3847/1538-4357/aab36a
- S. Brünken, L. Kluge, A. Stoffels, O. Asvany and S. Schlemmer, Astrophys. J. Lett. 783, L4 (2014). doi: 10.1088/2041-8205/783/1/L4
- S. Brünken, L. Kluge, A. Stoffels, J. Pérez-Ríos and S. Schlemmer, J. Mol. Spectrosc.332, 67–78 (2017). doi: 10.1016/j.jms.2016.10.018
- J.L. Doménech, S. Schlemmer and O. Asvany, Astrophys. J. 849, 60 (2017). doi: 10.3847/1538-4357/aa8fca
- D. Oepts, A. van der Meer and P. van Amersfoort, Infrared Phys. Technol. 36, 297–308 (1995). doi: 10.1016/1350-4495(94)00074-U
- M. Töpfer, T. Salomon, S. Schlemmer, O. Dopfer, H. Kohguchi, K. Yamada and O. Asvany, Phys. Rev. Lett. 121, 143001 (2018). doi: 10.1103/PhysRevLett.121.143001
- T. Salomon, M. Töpfer, P. Schreier, S. Schlemmer, H. Kohguchi, L.A. Surin, and O. Asvany, Phys. Chem. Chem. Phys. 21, 3440–3445 (2019). doi: 10.1039/C8CP04532A
- W.D. Allen, A.L.L. East and A.G. Császár, in Structures and Conformations of Nonrigid Molecules, edited by J. Laane, M. Dakkouri, B. van der Veken, H. Oberhammer (Kluwer, Dordrecht, 1993), pp. 343–373.
- A.G. Császár, W.D. Allen and H.F. Schaefer III, J. Chem. Phys. 108, 9751–9764 (1998). doi: 10.1063/1.476449
- M.J. Frisch, et al., Gaussian16 Revision B.01, 2016, Gaussian Inc. Wallingford CT.
- CFOUR, a quantum chemical program package written by J. F. Stanton, J. Gauss, M. E. Harding, P. G. Szalay with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, Y. J. Bomble, L. Cheng, O. Christiansen, M. Heckert, O. Heun, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, K. Klein, W. J. Lauderdale, D. A. Matthews, T. Metzroth, D. P. O'Neill, D. R. Price, E. Prochnow, K. Ruud, F. Schiffmann, W. Schwalbach, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J. D. Watts and the integral packages MOLECULE (J. Almlöf and P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H.-J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen. For the current version, see http://www.cfour.de.
- M. Kállay and P.R. Surján, J. Chem. Phys. 115, 2945 (2001). doi: 10.1063/1.1383290
- MRCC, a quantum chemical program suite written by M. Kállay, Z. Rolik, J. Csontos, P. Nagy, G. Samu, D. Mester, J. Csóka, B. Szabó, I. Ladjánszki, L. Szegedy, B. Ladóczki, K. Petrov, M. Farkas, P. D. Mezei, and B. Hégely. For the current version, see www.mrcc.hu.
- Z. Rolik, L. Szegedy, I. Ladjánszki, B. Ladóczki and M. Kállay, J. Chem. Phys. 139, 094105 (2013). doi: 10.1063/1.4819401
- T.J. Lee and P.R. Taylor, Int. J. Quant. Chem. Symp. 23, 199–207 (1989).
- R. Hellmann, E. Bich, and E. Vogel, Mol. Phys. 105, 3013–3023 (2007). doi: 10.1080/00268970701730096
- A.G. Császár, W.D. Allen, Y. Yamaguchi, and H.F. Schaefer III, , Ab initio determination of accurate ground electronic state potential energy hypersurfaces for small molecules, in Computational Molecular Spectroscopy (Wiley, New York, 2000), pp. 15–68.
- G. Tarczay, A.G. Császár, W. Klopper, V. Szalay, W.D. Allen and H.F. Schaefer III, J. Chem. Phys. 110, 11971–11981 (1999). doi: 10.1063/1.479135
- K. Aarset, A.G. Császár, E. Sibert, W.D. Allen, H.F. Schaefer III, W. Klopper and J. Noga, J. Chem. Phys. 112, 4053–4063 (2000). doi: 10.1063/1.481596
- A.G. Császár, M. Leininger and V. Szalay, J. Chem. Phys. 118, 10631–10642 (2003). doi: 10.1063/1.1573180
- A.G. Császár and A. Perczel, Progr. Biophys. Mol. Biol. 71, 243–309 (1999). doi: 10.1016/S0079-6107(98)00031-5
- F. Pollreisz, A. Gömöry, G. Schlosser, K. Vékey, I. Solt and A.G. Császár, Chem. Eur. J. 11, 5908–5916 (2005). doi: 10.1002/chem.200401226
- T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys. 106, 9639 (1997). doi: 10.1063/1.473863
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper and J. Olsen, Chem. Phys. Lett. 302, 437–446 (1999). doi: 10.1016/S0009-2614(99)00179-7
- H. Chung, B.J. Braams, K. Bartschat, A.G. Császár, G. Drake, T. Kirchner, V. Kokoouline and J. Tennyson, J. Phys. D: Appl. Phys. 49, 363002 (2016). doi: 10.1088/0022-3727/49/36/363002
- G. Tarczay, A.G. Császár, W. Klopper and H.M. Quiney, Mol. Phys. 99, 1769–1794 (2001). doi: 10.1080/00268970110073907
- N.C. Handy, Y. Yamaguchi and H.F. Schaefer III, J. Chem. Phys. 84, 4481 (1986). doi: 10.1063/1.450020
- D.R.G. Schleicher, D. Galli, F. Palla, M. Camenzind, R.S. Klessen, M. Bartelmann and S.C.O. Glover, Astron. Astrophys. 490, 521–535 (2008). doi: 10.1051/0004-6361:200809861
- S. Lepp and M. Shull, Astrophys. J. 280, 465–469 (1984). doi: 10.1086/162013
- G. Harris, A.E. Lynas-Gray, S. Miller and J. Tennyson, Astrophys. J. 617, L143–L146 (2004). doi: 10.1086/427391
- M. Telmini and C. Jungen, Phys. Rev. A 68, 062704 (2003). doi: 10.1103/PhysRevA.68.062704
- I. Bouhali, S. Bezzaouia, M. Telmini and C. Jungen, Phys. Rev. A 94, 022516 (2016). doi: 10.1103/PhysRevA.94.022516
- E.P. Hunter and S.G. Lias, J. Phys. Chem. Ref. Data 27, 413–656 (1998). doi: 10.1063/1.556018
- A.G. Császár, C. Fábri, T. Szidarovszky, E. Mátyus, T. Furtenbacher and G. Czakó, Phys. Chem. Chem. Phys. 13, 1085–1106 (2012). doi: 10.1039/C1CP21830A
- https://goldbook.iupac.org/, 2018.
- F. Paesani, R.E. Zillich and K.B. Whaley, J. Chem. Phys. 119, 11682–11694 (2003). doi: 10.1063/1.1624058
- Y. Kwon and K.B. Whaley, J. Low Temp. Phys. 140, 227–240 (2005). doi: 10.1007/s10909-005-6310-0
- D. Papp, A.G. Császár, K. Yamanouchi and T. Szidarovszky, J. Chem. Theory Comput. 14, 1523–1533 (2018). doi: 10.1021/acs.jctc.7b01136