
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We have studied the collision induced dissociation reactions of proton-bound diastereomeric adducts of S-1-phenylethanol and enantiomers of three different amino acids (tryptophan, phenylalanine, methionine). In all cases, the loss of S-1-phenylethanol from the adduct ion is the only observed process, and the relative abundance is found to be independent of the chirality of the amino acid. This is in contrast to earlier experiments on the dissociation of protonated tryptophan–2-butanol adducts, where chirality affected the results. Results obtained from quantum chemical computations support and provide a rationale for the experimental observations and highlight temperature as a possible factor of importance for the chiral effect in these types of systems.
GRAPHICAL ABSTRACT
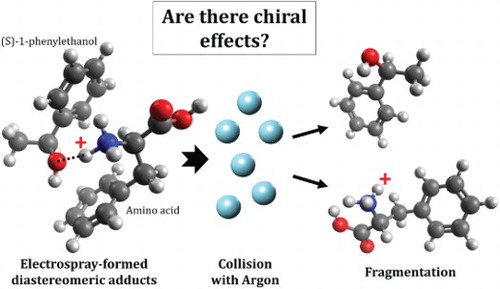
Introduction
The chemical and biological separation of enantiomers depends on chiral recognition involving weak molecular interactions. Although such interactions are in principle well understood, their relative strengths and the interplay between them is often a more complex issue [Citation1]. Because enantiomers require an anisotropic environment in order to disclose their differences, a chiral selector is in general required for their separation.
Chiral separation in the gas phase by means of ion mobility spectrometry combined with mass spectrometry (IMS-MS) was first demonstrated by Dwivedi et al. in 2006 [Citation2]. (R)- and (S)-2-butanol were added as modifiers to the IMS-MS system, and drift time separation of enantiomers from a racemic mixture was reported for a variety of compounds including methionine (Met). The authors explained their observation by the Pirkle three-point interaction rule [Citation3], which is a phenomenological description of the interaction of the chiral analyte with the anisotropic, chiral environment.
Unfortunately, it seems that more recent experiments have not fully reproduced the enantiomeric separation reported by Dwivedi et al.: differences in drift time were observed for enantiomers of various compounds when introduced separately, but separation did not occur for racemic mixtures [Citation4]. On the other hand, enantiomeric separation by IMS-MS using achiral modifiers in the drift cell has been observed, and was explained by the formation of transient alcohol clusters around the chiral ionic molecules which resulted in different collisional cross sections [Citation5,Citation6].
Kyluk et al. performed a series of experiments that essentially support the notion that the three-point interaction model may be an over-simplification [Citation7]. For interactions between enantiopure amino acids (tryptophan (Trp), methionine (Met), phenylalanine (Phe)) and selected chiral alcohols ((S)-2-butanol or (S)-1-phenylethanol), neither collisionally induced dissociation (CID) of the protonated amino acid, nor gas phase adduct formation between the protonated amino acid and the alcohol at 0.1 eV, showed any significant dependence on the stereochemical composition of the interacting partners. However, two caveats should be noted: firstly, the dissociation experiments were designed to occur under single-collision conditions, whereas IMS involves many collisions; and, secondly, that the random orientation of the ionic projectile and the neutral molecules during third-body stabilised adduct formation could have eliminated any inherent stereo-chemical selectivity.
An earlier study by the present authors on the proton-bound diastereomeric adducts of enantiomeric (R)- and (S)-2-butanol and (R)- and (S)-Trp indicated that the homo-chiral adducts are less stable towards collisionally induced dissociation than the hetero-chiral ones [Citation8]. Thus, in contrast to direct collisions between protonated Trp and chiral alcohols [Citation7], dissociation of intact adducts between protonated Trp and 2-butanol gives rise to stereo-chemical selectivity [Citation8].
For the sake of completeness, it should also be mentioned that in IMS-MS transition metals are often used to enhance enantiomeric separation by the formation of trimeric complexes [Citation9–13], a concept also exploited in other mass spectrometric applications [Citation14–17]. However, the specific metal–ligand interactions present in such complexes are not the focus of the current paper.
In order to better understand the nature of the interactions between the two chiral molecular entities within a protonated adduct and the dissociation dynamics of the latter upon collisional activation, we wanted to extend the scope of the previous study [Citation8] to a wider range of amino acids and alcohols. From a rather naïve perspective, replacing 2-butanol with the bulkier (S)-1-phenylethanol in the adducts could impose steric effects that could influence enantioselectivity. This paper therefore reports the results of CID experiments with the three previously studied amino acids (Trp, Met, Phe) and their adducts with (S)-1-phenylethanol. In addition to the experimental effort, we also report the results of complementary quantum chemical computations aimed at providing insight into the recognition mechanism.
Materials and methods
Experimental methods
The S- and R- forms of Trp, Met, Phe, and the S-form of 1-phenylethanol (>98% enantiopure) were purchased from Sigma Aldrich and used in experiments without further purification. A 0.1 mM solution of each amino acid was prepared in a mixture of water/methanol/acetic acid (49:49:2, v/v/v). 50 μL S-1-phenylethanol was then added to 10 mL of the mixture.
A hybrid quadrupole time-of-flight mass spectrometer (QTOF 2, Micromass/Waters, Manchester, UK) with an electrospray ion source, located at the Department of Chemistry, University of Oslo, was used for the experiments. The mass spectrometer was operated in tandem mass spectrometry mode (MS/MS). This mode enables an initial mass selection of the ion of interest and, following fragmentation induced by collision with Ar, mass identification of the dissociation products. The electrospray source was operated in the positive ion mode. The source temperature was 100°C, and the needle voltage was set to 3.2 kV. Proton bound complexes of amino acid and (S)-1-phenylethanol, spontaneously formed in the ion source, were mass selected by the quadrupole mass analyser and transferred to the hexapole collision cell. Here, interactions with argon took place at a nominal pressure of 1.0 × 10−4 mbar, i.e. under essentially single collision conditions with a collision energy of 0.5 eV in the center-of-mass frame (CoM). The energy distribution due to the Doppler effect is about 0.3 eV at full-width-at-half-maximum, and corresponds to ±35% of the nominal collision energy. However, this broadening appears not to effect the results presented here. The products of the reaction were transferred to the ToF section of the mass spectrometer, and registered by a microchannel-plate-based detector. A solution of water:methanol (50:50, v/v) was sprayed for 5–10 min after each measurement to clean the source. In order to avoid cross-contamination, for each enantiomeric form of amino acid, separate syringes and pipelines were used.
The combining, smoothing and integration of the recorded mass-spectra was undertaken using the MassLynx V4.0 software. The measurements for each amino acid were repeated several times. The fragment abundancies were normalised to the parent peak abundance and averaged over all repeated experiments.
Computational methods
The Avogadro advanced molecule editor [Citation18] was used for creating the initial molecular structures. The extra proton was located on the amine group in each amino acid [Citation19–21]. Molecular dynamics simulations (MD) were performed for each diastereomeric adduct using the density functional-based tight binding method DFTB+ (version 1.2) [Citation22]. The code is based on the SCC-DFTB approach [Citation23], and implements the second order expansion of the Kohn–Sham energy in terms of charge density fluctuation [Citation24,Citation25]. This first round of computations provided the structures with corresponding potential energies, calculated for each step of the MD simulation.
The lowest energy structures were taken as the initial geometry for further computations. The geometry and energy optimisation of the obtained structures were carried out applying density functional theory methods (DFT) available in the GAUSSIAN 09 package [Citation26]. The hybrid B3LYP correlation functional [Citation27,Citation28] was adapted in a first series of computations using a basis set with extra polarisation and diffuse functions (6-311++G**). This method has shown good accuracy in predicting hydrogen bonding and other weak interactions, and has been successfully used for conformational analysis of amino acids [Citation29–32]. To include correction for dispersion, the Grimme’s D3 scheme with Becke–Johnson damping was incorporated in the computations [Citation33–36]. The relative stability of the complexes is described in terms of relative Gibbs energies computed at different temperatures using the lowest energy structures, applying the rigid rotor/harmonic oscillator approximation to estimate spectroscopic entropy and heat capacity.
Experimental results
Under the electrospray conditions pertaining to these experiments, the amino acids under investigation readily formed proton bound adducts with (S)-1-phenylethanol, without giving rise to higher order complexes.
The top panels in Figure (a–c) show plots of the mass-spectra recorded from the CID of the following adducts: (S)-TrpH+/(S)-1-phenylethanol, (S)-PheH+/(S)-1-phenylethanol, and (S)-MetH+/(S)-1-phenylethanol, respectively. The data in each figure have been normalised such that the parent-ion peak has an intensity of 1.0. To investigate the enantioselectivity of the dissociation reaction, the corresponding adducts formed using the R-forms of the same amino acids were also studied; these are shown in the bottom panels of Figure (a–c) (where these data are plotted upside down).
Figure 1. Relative abundance of the protonated amino acids obtained in one of consequent experiments from collisions of (a) (S/R)-TrpH+/(S)-1-phenylethanol, (b) (S/R)-PheH+/(S)-1-phenylethanol, and (c) (S/R)-MetH+/(S)-1-phenylethanol with argon at 0.5 eV collision energy (CoM). The spectra are normalized so that the parent ion intensity is 1.00.
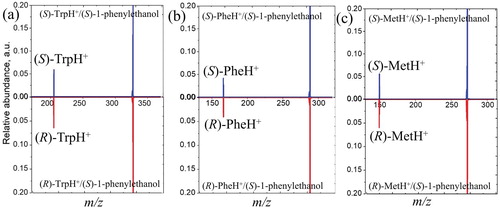
In all cases, the only product ion observed from the collision is the protonated amino acid. No other fragments were detected, also indicating that the collisions deposited insufficient energy in the ionic fragment to induce further dissociation.
The observed single dissociation channel indicates that the interaction between the two entities in the adduct is in all cases rather weak. The observation of protonated amino acid products, as opposed to the protonated alcohol, is in good agreement with the generally accepted higher proton affinity of these amino acids compared with that of (S)-1-phenylethanol. In this respect, the present results are consistent with our previous experiments with 2-butanol [Citation8].
Detailed data analysis has been undertaken to evaluate the stereoselectivity in these fragmentation reactions. The relative abundance of the fragment obtained in CID of the homo- and hetero-chiral adducts was compared. In the first set of CID experiments, homo-chiral adducts with Phe and Met appeared to have a slight tendency to be less stable than the corresponding hetero-chiral adducts. However, averaging the results over all experiments in the series, the difference between the relative abundance of fragments obtained from homo- and hetero-chiral adducts is within a 2σ interval and shows chirally independent dissociation. We therefore conclude that (S)-1-phenylethanol as a proton bound adduct does not give rise to any significant chiral recognition for any of the amino acids considered here, in contrast to our previous result with Trp and 2-butanol, [Citation8].
Model computations
From the computations we were able to identify the most stable structure for each diastereomeric adduct. In addition to applying B3LYP-D3/6-311++G** to the lowest lying structures from the MD approach described above, we also probed the stability of likely candidate structures not in the MD set to ensure that the lowest energy structure had been identified in each case. All adducts have similar intermolecular bonding by having a hydrogen bond between the protonated amino group of the amino acid and the oxygen in the hydroxyl group of the alcohol (Figure ).
Figure 2. The most stable structures calculated with B3LYP-D3/6-311++G** for the homo-chiral (left) and hetero-chiral (right) proton bound adduct of (a) Trp and (S)-1-phenylethanol; (b) Met and (S)-1-phenylethanol; (c, d) Phe and (S)-1-phenylethanol.
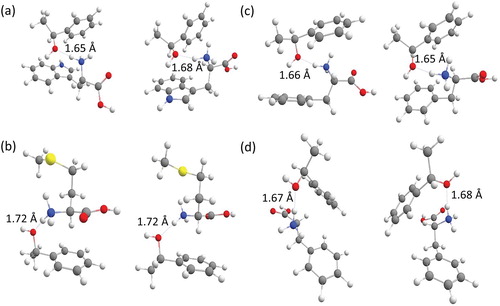
TrpH+/(S)-1-phenylethanol adducts are represented by a single most stable conformer for each chirality of Trp (Figure (a)). Met containing diastereomeric adducts also have a single most populated conformation (Figure (b)). The adducts with Phe have two different conformations that are almost equally populated (Figure (c,d)).
(S)- and (R)-TrpH+/(S)-1-phenylethanol have very similar structures, having NH…O bond lengths of 1.65 and 1.68 Å, respectively. In both cases, we observe that the hydrogen of the hydroxyl group of the alcohol forms a hydrogen bond to the π-system of the aromatic ring of Trp. A similar type of hydrogen bond is also seen to exist between the protonated amino group of Trp and the aromatic ring of 1-phenylethanol, in both cases (Figure (a)).
Met containing diastereomeric adducts have intermolecular NH…O bond lengths of 1.72 Å for both enantiomeric amino acids. The amino acid sidechain points away from the 1-phenylethanol entity, and is not involved in the intermolecular interaction (Figure (b)).
The diastereomeric adducts with Phe exist in two stable conformations of comparable energy. The main difference between them is the relative orientation of the aromatic rings, which are either far away from each other (Figure (c)) or point towards each other (Figure (d)). The conformations with the remote orientation of the rings have NH…O bond lengths of 1.66 and 1.65 Å for the homo- and hetero-chiral complex, respectively; for both diastereomeric forms, the hydroxyl group of 1-phenylethanol forms a hydrogen bond to the aromatic ring of Phe. In the conformations with the rings in proximity to each other, the NH…O bond lengths for the homo-chiral and hetero-chiral structures are 1.67 and 1.68 Å, respectively.
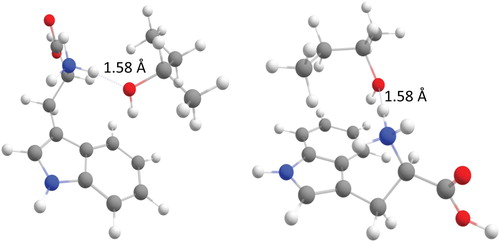
In order to compare the results obtained in this study with the previously reported work [Citation8], the structures from the previous study ((R)- and (S)-TrpH+/(S)-2-butanol) were reoptimized using B3LYP-D3/6-311++G** (Figure ). The relative position of the 2-butanol and Trp entities is comparable for both chiral forms, with 2-butanol located in proximity to the indole ring of the amino acid, and with a hydrogen bond between the OH group of the alcohol and the aromatic ring of the amino acid. An NH…O(H) hydrogen bond ties the protonated amino group of Trp to the hydroxyl group of the alcohol, as in the adducts with 1-phenylethanol discussed above. The intermolecular hydrogen bond length is 1.58 Å for both adducts. Some structural differences can, however, be pointed out. In the hetero-chiral adduct (Figure , right), 2-butanol is located directly above the indole ring, whereas in the homo-chiral adduct (Figure , left) 2-butanol is only interacting with the side of the ring. This difference could explain why the hetero-chiral adduct is more stable than the homo-chiral (see below).
Figure 3. The most stable structures calculated at the B3LYP-D3/6-311++G** for homo-chiral (left) and hetero-chiral (right), proton bound adduct of TrpH+/(S)-2-butanol.
The stability of the all diastereomeric adducts covered here have been compared by computing the Gibbs energy of adduct formation for each pair in question, i.e. for reactions of the type
(1)
(1) where amac and alc are the amino acid and alcohol, respectively. Based upon this, the difference in relative Gibbs energy for the homo- and hetero-chiral diastereomers were computed as a function of the ion temperature in the range 25–500 K (Figure ).
Figure 4. Computed difference in the Gibbs free energy of adduct formation for the homo- and hetero-chiral adducts of TrpH+/(S)-1-phenylethanol (blue), MetH+/(S)-1-phenylethanol (red), PheH+/(S)-1-phenylethanol (green) and TrpH+/(S)-2-butanol (black). A positive value indicates that the hetero-form is the more stable one.
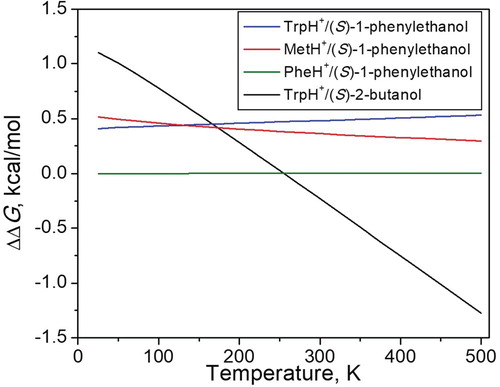
The hetero-chiral adduct of TrpH+/(S)-1-phenylethanol is more stable than the homo-chiral by approximately 0.5 kcal/mol at all temperatures, and this difference increases only slightly with temperature (Figure , blue line). The hetero-chiral adduct of MetH+/(S)-1-phenylethanol is also more stable than the homo-chiral by approximately 0.4 kcal/mol, though this difference decreases slightly with temperature (Figure , red line). The most stable conformations of (S)-PheH+/(S)-1-phenylethanol and (R)-MetH+/(S)-1-phenylethanol have no significant differences, and show similar stability over the whole temperature range (Figure , green line).
(S)- or (R)-TrpH+/(S)-2-butanol shows a quite different behaviour from those above. The difference in Gibbs energy between the homo- and hetero-chiral structures is 1.1 kcal/mol at 25 K, with the hetero-chiral complex being the more stable. However, the energy difference decreases with increasing temperature and reaches 0 at 255 K. With a further increase in temperature, the homo-chiral adduct becomes more stable with ΔΔG = −1.3 kcal/mol at 500 K (Figure ). The most significant contributions to the different entropy terms in the diastereomeric complexes can be explained by differences in the lowest energy vibrations. However, great care should be taken in the interpretation of these values, since these modes turn out to be hindered rotors rather than harmonic vibrations. For example, for homo-chiral TrpH+/(S)-2-butanol, the hindered rotation of the (S)-2-butanol entity around the C–O bond at 14 cm−1 gives the largest contribution to the entropy. This mode is not present for the hetero-chiral R-TrpH+/(S)-2-butanol, for which the lowest frequency mode at 30 cm−1 stems from a collective vibrational mode that involves both TrpH+ and 2-butanol. Despite these caveats, it seems clear that the 2-butanol complexes in general are less tightly bonded, as they lack stabilising hydrogen bonds to π-systems when compared to the complexes with 1-phenylethanol.
Discussion and conclusion
The process of CID can be separated into two steps; the collision of the adduct with an argon atom (fast) and the subsequent dissociation into the products (the constituent molecular entities, slow):
(2)
(2) For a given diastereomeric pair, we note that the products formed from the hetero- and homo-chiral adducts have the same energy, while the energies of the reacting adducts are slightly different. If we assume that the energy distribution of [amacH+(alc)]* is the same in both cases, the kinetics of the second step can in principle be simulated using statistical rate theory (Arrhenius equation, transition state theory or RRKM theory). In the simplest form, we could assume the relationship between the rate coefficients for the two processes to be,
(3)
(3)
The effective temperature can then be roughly estimated from the original temperature (for electrospray ionisation this is probably around 150 K [Citation37]) of the adduct plus the collision energy. For the sake of argument we could set the effective temperature to, 2000 K. An energy difference of 0.5 kcal/mol, relevant for some of the 1-phenylethanol adducts studied here, would then be equivalent to , a value close to the limit of significant detection of enantioselectivity in our experiments. The same argument may apply to adducts formed in ion–molecule reactions, as in the previous study by Kyluk et al. [Citation7], although those experiments were probably conducted at lower effective temperatures, which would enhance enantioselectivity. Despite this, enantioselectivity was not detected in those experiments either. A qualitative discussion of some of these aspects were given in the extensive review of Zehnacker [Citation38].
As already mentioned, in our earlier study [Citation8] it was found that the diastereomeric adducts of protonated Trp and 2-butanol could be differentiated by CID. In contrast, CID of the amino acid (Trp, Met and Phe) and 1-phenylmethanol adducts investigated in this work did not result in any chiral recognition. This can be found somewhat counterintuitive, as the bulkier (S)-1-phenylethanol might be expected to impose steric effects that could further enhance enantioselectivity, in comparison to 2-butanol. The matter is elucidated by quantum chemical computations.
For the homo- and hetero-chiral adducts of Trp and 2-butanol, the computational work herein shows that there is a difference in the Gibbs energy of formation for the two diastereomeric complexes. In addition, this energy difference is temperature dependent, and, as a consequence, which complex is more stable might change over the temperature range. However, it should be emphasized that the entropy effect detected depends on a crude statistical model.
For the protonated adducts of Trp, Met and Phe with (S)-1-phenylethanol, the computed Gibbs energy differences for the diastereomers were found to be smaller and showed little temperature dependence over the range 25–500 K. This arises as a consequence of the lowest energy structures for two diastereomeric adducts of a specific amino acid, which turns out to be very similar with the same type of intermolecular interaction via a hydrogen bond between protonated amino group and the oxygen in the hydroxyl group of the alcohol. In addition, there is a stabilising interaction between the hydrogen in the hydroxyl group and the π−system of the amino acid's aromatic ring (if present).
The structures of the diastereomeric adducts of TrpH+ with (S)-2-butanol are also similar to each other, with identical bond lengths for the hydrogen bond linking the two molecular entities together. The OH-group/π−system interaction referenced above exists also in the adducts of TrpH+ with (S)-2-butanol. However, differences in structure between the two diastereomers affects the position of 2-butanol relative to the aromatic ring, which is likely relevant for the difference in Gibbs energy.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
O. Rebrov http://orcid.org/0000-0002-4940-0481
M. J. Ryding http://orcid.org/0000-0003-2693-4939
E. Uggerud http://orcid.org/0000-0003-2732-2336
Additional information
Funding
References
- A. Berthod, in Chiral Recognition in Separation Methods, edited by A. Berthod (Springer, Berlin, Heidelberg, 2010).
- P. Dwivedi, C. Wu, L.M. Matz, B.H. Clowers, W.F. Siems and H.H. Hill, Anal. Chem. 78, 8200–8206 (2006). doi: 10.1021/ac0608772
- W.H. Pirkle and T.C. Pochapsky, Chem. Rev. 89, 347–362 (1989). doi: 10.1021/cr00092a006
- R. Fernandez-Maestre, J. Mass Spectrom. 53, 598–613 (2018). doi: 10.1002/jms.4190
- H.K. Holness, Ph.D. Dissertation, Florida International University (2013).
- H.K. Holness, A. Jamal, A. Mebel and J.R. Almirall, Anal. Bioanal. Chem. 404, 2407–2416 (2012). doi: 10.1007/s00216-012-6365-0
- K. Kulyk, O. Rebrov, M. Ryding, R.D. Thomas, E. Uggerud and M. Larsson, J. Am. Soc. Mass Spectrom. 28, 2686 (2017).
- O. Rebrov, K. Kulyk, M. Ryding, R.D. Thomas, E. Uggerud and M. Larsson, Chirality. 29, 115–119 (2017). doi: 10.1002/chir.22679
- A. Mie, M. Jörnten-Karlsson, B.O. Axelsson, A. Ray and C.T. Reimann, Anal. Chem. 79, 2850–2858 (2007). doi: 10.1021/ac0618627
- A. Mie, A. Ray, B.O. Axelsson, M. Jörnten-Karlsson and C.T. Reimann, Anal. Chem. 80, 4133–4140 (2008). doi: 10.1021/ac702262k
- V. Domalain, M. Hubert-Roux, V. Tognetti, L. Joubert, C.M. Lange, J. Rouden and C. Afonso, Chem. Sci. 5, 3234–3239 (2014). doi: 10.1039/C4SC00443D
- X. Yu and Z.P. Yao, Anal. Chim. Acta. 981, 62–70 (2017). doi: 10.1016/j.aca.2017.05.026
- J.R. Enders and J.A. McLean, Chirality. 21, E253–E264 (2009). doi: 10.1002/chir.20806
- W.A. Tao and R.G. Cooks, Anal. Chem. 75, 25A (2003). doi: 10.1021/ac0312110
- L. Wu, F.G. Vogt and J. Pharm, Biomed. Anal. 69, 133–147 (2012). doi: 10.1016/j.jpba.2012.04.022
- H. Awad and A. El-Aneed, Mass Spectrom. Rev. 32, 466 (2013).
- X. Yu and Z.P. Yao, Anal. Chim. Acta. 968, 1–20 (2017). doi: 10.1016/j.aca.2017.03.021
- M.D. Hanwell, D.E. Curtis, D.C. Lonie, et al., J. Cheminform. 4, 17 (2012). doi: 10.1186/1758-2946-4-17
- H. Lioe, R.A.J. O’Hair and G.E. Reid, J. Am. Soc. Mass. Spectrom. 15, 65–76 (2004). doi: 10.1016/j.jasms.2003.09.011
- H. El Aribi, G. Orlova, A.C. Hopkinson and K.W.M. Siu, J. Phys. Chem. A. 108, 3844–3853 (2004). doi: 10.1021/jp0374915
- F. Rogalewicz, Y. Hoppilliard and G. Ohanessian, Int. J. Mass Spectrom. 195–196, 565–590 (2000). doi: 10.1016/S1387-3806(99)00225-0
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A. 111, 5678–5684 (2007). doi: 10.1021/jp070186p
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev B. 58, 7260–7268 (1998). doi: 10.1103/PhysRevB.58.7260
- P. Koskinen and V. Makinen, Comput. Mater. Sci. 47, 237–253 (2009). doi: 10.1016/j.commatsci.2009.07.013
- G. Seifert and J.O. Joswig, Comput. Molec. Sci. 2, 456–465 (2012). doi: 10.1002/wcms.1094
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, et al., Computer code Gaussian 09, Revision C.01 (Gaussian, Inc., Wallingford, CT, 2010).
- A.D. Becke, J. Chem. Phys. 98, 5648–5652 (1993). doi: 10.1063/1.464913
- C. Lee, W. Yang and R.G. Parr, Phys. Rev. 37, 785–789 (1988). doi: 10.1103/PhysRevB.37.785
- B. Lambie, R. Ramaekers and G. Maes, J. Phys. Chem. A. 108, 10426–10433 (2004). doi: 10.1021/jp047192v
- M. Noguera, L. Rodriquez-Santiago, M. Sodupe and J. Bertran, J. Mol. Struct. 537, 307–318 (2001). doi: 10.1016/S0166-1280(00)00686-2
- S.G. Stepanian, I.D. Reva, E.D. Radchenko, M.T.S. Rosado, M.L.T.S. Duarte, R. Fausto and L. Adamowicz, J. Phys. Chem. A. 102, 1041–1054 (1998). doi: 10.1021/jp973397a
- S.G. Stepanian, I.D. Reva, E.D. Radchenko and L. Adamowicz, J. Phys. Chem. A. 102, 4623–4629 (1998). doi: 10.1021/jp973479z
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys. 132, 154104 (2010). doi: 10.1063/1.3382344
- A.D. Becke and E.R. Johnson, J. Chem. Phys. 123, 154101 (2005). doi: 10.1063/1.2065267
- E.R. Johnson and A.D. Becke, J. Chem. Phys. 123, 024101 (2005). doi: 10.1063/1.1949201
- E.R. Johnson and A.D. Becke, J. Chem. Phys. 124, 174104 (2006). doi: 10.1063/1.2190220
- A.E.K. Sunden, K. Støchkel, S. Panja, U. Kadhane, P. Hvelplund, S.B. Nielsen, H. Zettergren, B. Dynefors and K. Hansen, J. Chem. Phys. 130, 224308 (2009). doi: 10.1063/1.3149784
- A. Zehnacker, Int. Rev. Phys. Chem. 33, 151–207 (2014). doi: 10.1080/0144235X.2014.911548