

Abstract
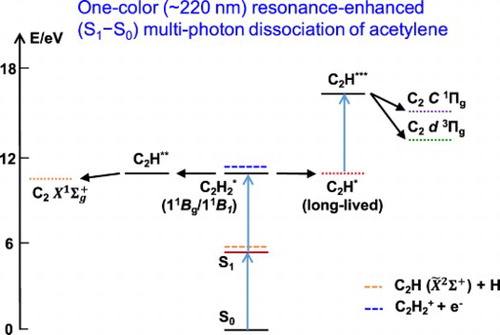
In a recent paper, we demonstrated that one-colour (∼220 nm), resonance-enhanced (SS
), photodissociation of acetylene generates strong
Swan band (
) and
Deslandres-d'Azambuja band (
) fluorescence, and long-lived (>3 µs) fluorescence from an electronically-excited
H
species. It was not known whether the
and
states are also directly populated in this process. In this work, multiple vibration-rotation transitions between the
-state v = 2 and the X-state v = 0 level are examined by time-resolved frequency-modulation (FM) spectroscopy. The photolysis laser wavelength is tuned into resonance at the one-photon level with S
S
transitions that populate individual rotational levels of the S
-conformer
,
, and
vibrational states. By comparing the phase of the FM signals from the
transitions with that from the Rb D
-line absorption transition, we determine that, for all of the probed A−X transitions, the X-state level is more populated than the A-state level. We propose that the acetylene S
level is excited by the second photon to an acetylene dissociation precursor state, which undergoes sequential C-H bond-breaking to produce the
state. The dissociation precursor is assigned as the
valence state, which correlates to a doubly-excited configuration,
, at linear geometry. Based on the rotational distributions of the
-state fragments, we believe that at least one of the transition states involved in the photolysis via S
has a larger CC-H bend-angle for the departing H-atom than that involved in the S
and
photolysis.
GRAPHICAL ABSTRACT
Acknowledgments
The authors are grateful to the anonymous reviewer for suggesting the doubly-excited state as a possible assignment for the super-excited acetylene precursor state at the two-photon level. The authors also thank Dr. Gregory Hall from Brookhaven National Laboratory for the loan of multiple FM components, and provided useful advice on the FM setup. This material is based upon work supported by the U.S. Department of Energy, Office of Science, Chemical Sciences, Geosciences, and Biosciences Division of the Basic Energy Sciences Office, under Award Number DE-FG0287ER13671. The Ti-sapphire laser and the solid-state 532 nm pump laser are provided by the Air Force Office of Scientific Research, under grant number FA9550-16-1-0117. The manuscript preparation was carried out at Lawrence Livermore National Laboratory under the auspices of the U.S. Department of Energy under Contract DE-AC52-07NA27344.
Disclosure statement
No potential conflict of interest was reported by the author(s).
ORCID
Jun Jiang http://orcid.org/0000-0002-3526-3797
Zhenhui Du http://orcid.org/0000-0002-3102-8773
Robert W. Field http://orcid.org/0000-0002-7609-4205