
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Singlet fission is a potential mechanism to enhance the performance of current solar cells. However, the actual mechanism is still a matter of debate, with charge transfer states believed to play an essential role. The probability of the overall process can be related to the electronic coupling between the electronic states. Here, we explore the excited states of three pairs of tetracene with different relative orientation in the crystal structure showing different electronic couplings and identify the role of charge transfer states. First, a suitable theoretical method for the study of the tetracene pairs is determined by comparing time-dependent density functional theory with wave function-based methods in terms of excitation energies, so-called exciton descriptors, and graphical tools such as electron-hole correlation plots and natural transition orbitals. The results show the presence of low-lying charge transfer states in those tetracene pairs with non-zero electronic coupling, suggesting a superexchange-mediated mechanism, and high-lying charge resonance states for the pair with zero electronic coupling. Finally, the lower electron-hole correlation coefficients for pairs with non-zero coupling speak in favour of the superexchange-mediated mechanism, as a weaker Coulombic attraction due to the mixing with charge transfer states further facilitates the formation of the state from the photoexcited molecule.
GRAPHICAL ABSTRACT
1. Introduction
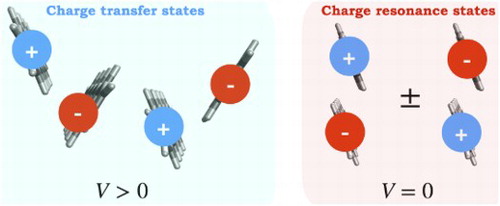
Predictions point out that if the temperature of the Earth raises above four degrees within the next years, around 43 to 58% of the current species inhabiting the planet will disappear [Citation1]. Therefore, clean and renewable ways to generate electricity are needed. The conversion of sunlight into electricity seems to be one of the most promising ways to face the current environmental challenges [Citation2]. Nevertheless, the artificial light-harvesting is still problematic due to the low quantum yields shown in solar cells [Citation3]. Therefore, ways to improve their performance are needed. Singlet fission (SF) is a multiexcitonic generation process that occurs in organic solids, [Citation4, Citation5] during which two coupled triplets are formed from a singlet photoexcited system in an overall spin-allowed process (see Figure ) [Citation4, Citation5]. SF has been explored as an alternative process to enhance the current performance of solar cells and break the so-called Shockley–Queisser theoretical limit of 34% [Citation6, Citation7], since in principle two pairs of charge carriers can be generated per absorbed photon. It has been estimated that the inclusion of a SF material layer to a single-junction photovoltaic device could increase its efficiency from 34% to approximately 45% if the charge carriers are effectively harvested [Citation8]. Furthermore, simulations have predicted that the addition of a SF material could increase the efficiency of a current silicon-based solar cell by up to 4.2% [Citation9]. Nevertheless, the low number of molecules exhibiting SF and the poor understanding of how this process takes place have limited the development of SF-based solar cells. During recent years, experimental works have started to demonstrate the potential of SF materials for enhancing the performance of solar cells within a variety of architectures [Citation10, Citation11].
Figure 1. Singlet fission process occurring via three different mechanisms: a molecule is excited upon absorption of a photon, then the excited molecule transfers part of its energy to a neighbour to form two triplets. = ground state,
= lowest singlet, T = triplet,
= cation and
= anion.
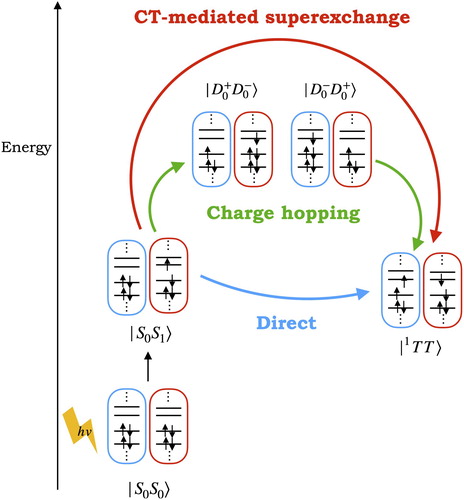
Theoretical studies have been performed in order to find new SF molecules that fulfill the critical energetic criteria , [Citation12, Citation13] and also to understand its underlying mechanism (a detailed review can be found in Ref. [Citation14]) but many questions of how this process actually occurs are still active areas of research; these include the role of the vibrational modes [Citation15–19] and charge transfer (CT) states [Citation20, Citation21] in the SF process.
Generally, the simple picture in which the two coupled triplets () are formed from the local excitations (
and
) via a direct two-electron transfer is accepted (blue arrow in Figure ). However, CT states (
and
) have been believed to play an essential role in the conversion to the
state; if the energy of these states is close to the energy of the
state a charge hopping [Citation22] mechanism can occur, in which the CT states are populated and the process occurs involving them (green arrow in Figure ), but if the CT state energies are higher than
, then a CT-mediated superexchange [Citation22] process might occur where CT states mix with the initial and final states facilitating the conversion (red arrow in Figure ). Thus, it is crucial to determine the energetic location of CT states to identify the underlying mechanism of the SF process.
The identification, characterisation and investigation of the excited states of a molecule and their relation with the SF process can be aided by quantum chemical calculations. Recently a series of excited state analysis tools (also known as exciton descriptors) based on the one-electron transition density matrix (1TDM) and its molecular orbital representation have been developed [Citation23, Citation24]. In this work a subset of exciton descriptors based on the 1TDM as well as graphic tools such as electron-hole correlation plots and natural transition orbitals, were used to analyse the excited states of tetracene, a widely known molecule that experimentally exhibits SF [Citation17, Citation18, Citation20, Citation25–29], in its monomeric, dimeric and trimeric forms. Our results provide detailed insights into the role of the CT states in the SF process for the pairs of tetracene molecules.
The paper is organized as follows: a subset of the exciton descriptors and how they can be applied to identify the character of an excited states are described in Section 2.1. In Section 2.2 calculation of electronic couplings, which is proportional to the rate of SF process, is discussed using a non-orthogonal configuration interaction approach. Then, in Section 4.1 we compare the performance of linear response time-dependent density functional theory [Citation30] (TD-DFT) against wave function-based methods using a tetracene molecule in order to determine a suitable functional for the description of the tetracene pairs. In Section 4.2 the excited states of three tetracene pairs with different relative orientation and electronic couplings are characterised based on the exciton descriptors and the role of the CT and charge resonance states in the SF process is analysed. Our concluding remarks are given in Section 5.
2. Theory
2.1. Exciton analysis and exciton descriptors
The central property in the excited state analysis is the 1TDM between the ground () and excited (
) state [Citation23]. The 1TDM offers a compact description of an electronic transition, i.e. it describes concisely the electron-hole pair formed upon absorption of a photon [Citation31]. The matrix representation in an orbital basis set {
} of the 1TDM takes the form [Citation32]
(1)
(1) where
and
represent the μ and ν orbitals where the positively-charged electron-hole and the negatively-charged excited electron are located, respectively. The 1TDM element
is defined as
, where
and
are the one-particle creation and annihilation operators, respectively.
can be then used to compute the so-called exciton descriptors that allow to identify, characterise and investigate the nature of the excited states of a molecule. The details of how the descriptors are computed from the 1TDM are beyond the scope of this work but the reader is referred to Ref. [Citation23] for further details. In the following, a short overview of a subset of the exciton descriptors and how to apply them in an excited state analysis is provided. The characterisation of the excited states based on the above mentioned tools has been discussed in more details previously, [Citation33] and for a more detailed discussion on the theory behind this excited state analysis the interested reader is referred to Ref. [Citation23, Citation24, Citation31].
One of the analysis that can be done on the 1TDM is to partially integrate its square while restricting the hole to a fragment A and the electron to a fragment B, [Citation34] represented as
(2)
(2)
are the so-called charge-transfer numbers and represent the probability of finding the hole on fragment A while the electron is on fragment B, [Citation23] or in practical terms indicate from where to where the electron density is transferred during the excitation (which could be pairs or group of atoms). A useful way to analyse
, particularly when more than two fragments are defined, are the so-called electron-hole correlation plots. These plots are generated by partitioning the system into fragments and computing the
between them; the resulting values are visualized as a pseudo-colour matrix with size
where n is the number of fragments [Citation34]. Local excitations correspond to the diagonal of the matrix going from lower left to upper right, [Citation35] whereas CT contributions occurring between the fragments are indicated in the off-diagonal elements. Additionally to the visualization of
, the off-diagonal contributions can be summed up to obtained the so-called total charge transfer (
) number defined as [Citation36]
(3)
(3) with Ω being the squared (Frobenius) norm
(4)
(4) Ω indicates the single excitation character of an excited state and its value is typically in the range between zero and one, where one indicates a pure single excitated state [Citation34]. For TDA-TDDFT, used in this paper, the value of Ω is always one, by definition [Citation37]. The values of
go from zero to one, in which zero corresponds to a local excitation and one to a completely separated CT state [Citation32]. These descriptor is useful to preliminarily determine if an excited state corresponds to a local excitation or a CT state, however such assignment shall be done analysing more than one exciton descriptor as it will be discussed later.
As stated above, the first step is to determine the character of a particular excited state, i.e. if it corresponds to a local excitation, to a Rydberg states or a CT state. The identificacion of CT states is rather challenging from the theoretical point of view, and it is important to differentiate between two types: (1) CT states in which there is a permanent shift of the electron density, and (2) charge-resonance (CR) states in which no net charge transfer occurs [Citation33]. The identification of these states could be confusing, but CT and CR states can be distinguished by measuring the distance between the centroids of the electron and hole expressed as [Citation33]
(5)
(5) where
and
are the positions of the centroids of the electron and hole, respectively. If for an excited state
this can be identified as a CT state, whereas
might indicate either a CR state or a local excitation. In order to confirm if the state corresponds to a CR or a local excitation, the so-called exciton size (
) can be used which measures the root-mean-square separation distance between the hole and the electron taking into consideration their spatial distributions [Citation33].
is defined as
(6)
(6) Their values are rather different for local excitations and CR states as described later. Such difference can only be seen when comparing the corresponding
values of the excited states for a specific system.
The next two descriptors described in this work correspond to the electron and hole sizes denoted as and
, respectively, and expressed as [Citation33]
(7)
(7) and
(8)
(8) These quantities represent the spatial distribution of the hole and the electron with respect to their centroids.
and
are useful to differentiate between types of excited states where
and
are rather different, e.g. Rydberg states, core-excitations, etc., with
the key quantity to differentiate Rydberg from valence states [Citation31]. The last descriptor presented in this work is the so-called electron-hole correlation coefficient (
) defined as
(9)
(9) where
, which describes the spatial relation between the electron and hole. Its values go from -1 to +1, and the sign of
gives information about the joint exciton; negative values indicate that the electron and hole avoid each other in space. On the opposite, positive values suggest that the exciton is bound by electrostatic interaction, and if
there is no electron-hole correlation [Citation38].
Another useful graphic tool in the description of an excited state are the so-called natural transition orbitals (NTOs). The NTOs result from a single-value decomposition of the 1TDM and give a compact and state-specific description of the excitation [Citation23]. With having all these tools in hand, one can go beyond the classical approach of visualizing orbitals by analysing the exciton descriptors described above.
2.2. Electronic couplings
The probability of SF to occur in a system can be approximated by means of the Fermi's golden rule [Citation39]:
(10)
(10) where
represents the electronic coupling between two (diabatic) states, i and f, and
denotes the density of states per energy E unit. Electronic couplings have been evaluated with different approaches, [Citation14, Citation40] but in this work a non-orthogonal configuration interaction (NOCI) approach is employed, which is interesting for the SF process since the interaction between the states can be calculated directly [Citation13, Citation41].
In the NOCI approach, wave functions for the monomers in a cluster of N molecules describing the ,
,
,
and
states are generated. These molecular wave functions can be chosen to be of the CASSCF-type. In this work, the study is restricted to pairs of molecules. Then, many-electron basis functions (MEBFs) representing the following electronic states of the pair of molecules are generated as antisymmetrized products of the molecular wave functions:
,
,
,
,
and
. Hamiltonian and overlap matrix elements between the MEBFs are calculated, and the electronic coupling is obtained according to [Citation42]
(11)
(11) where i and f are the initial and final states of interest, respectively, which in the case of SF correspond to the photoexcited monomers (
and
) and the singlet-coupled triplet state (
).
3. Computational details
Ground state optimisation was carried out at the ωB97X-D/ANO-S-VDZP level of theory, including Grimme's dispertion correction [Citation43]. Excited state calculations were carried out using TD-DFT within the Tamm–Dancoff approximation with a variety of functionals: BLYP, B3LYP, BHLYP, CAM-B3LYP and ωB97X-D. Additionally, two variants of the algebraic diagrammatic construction of second order [Citation44] (ADC(2)-s and ADC(2)-x) and equation-of-motion coupled-cluster singles and doubles (EOM-CCSD) were used. The methods were employed with the 6-31G, 6-311G, cc-pVDZ, cc-pVTZ and ANO-S-VDZP basis sets. All calculations mentioned above were performed with the Q-Chem 5.1 quantum chemistry package [Citation45]. Pairs of tetracene molecules were identified and taken from the optimised crystal structure (see Supporting Information for crystal structure optimization). Excited state analysis was performed on the pairs of tetracene at the TD-DFT/ANO-S-VDZP level of theory employing CAM-B3LYP and ωB97X-D. Cartesian coordinates of all the relevant structures are given in the Supporting Information.
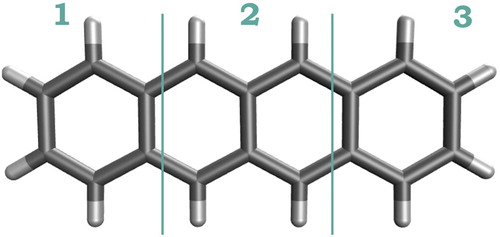
Figure shows the symmetric fragmentation of tetracene used in this work; three different fragments resulted with two benzene-like rings in the outer and a six-carbon fragment in the middle. Based on this fragmentation, the electron-hole correlation plots for the four lowest singlet excited states of the pairs are obtained at the CAM-B3LYP/ANO-S-VDZP and ADC(2)-s/ANO-S-VDZP levels of theory. Post-processing of results and electron-hole correlation plots were generated with the TheoDORE 1.7.2 analysis package [Citation35, Citation46]. In this work we use a grey scale for the electron-hole correlation plots with black corresponding to the highest and white to zero (or lowest
).
Figure 2. Labelled fragmentation scheme of the tetracene molecule used for the calculation of and construction of the electron-hole correlation plots.
For the electronic coupling calculations, the molecular wave functions for the pairs of tetracene molecules were generated at the CASSCF(4,4)/cc-pVDZ level of theory using the GAMESS-UK package [Citation47]. Then, the protocol outlined in Ref. [Citation13] was employed to obtained the diabatic states. The GronOR package [Citation41] was used to compute the Hamiltonian and overlap matrix elements between the states. Electronic couplings were calculated as defined in Section 2.2, allowing the mixing of the MEBFs describing the CT states to the and
states.
4. Results and discussion
4.1. Tetracene molecule
Table collects the excitation energies and oscillator strengths (f) for the four lowest singlet excited states of tetracene molecule calculated with TD-DFT (various functionals) as well as wave function based methods using ANO-S-VDZP basis set. In previous tetracene study [Citation48], the ANO-S-VDZP basis set was found to have a superior performance than other polarised double-ζ basis sets in the excitation energies, wave function overlaps and matrix-based descriptors. Influence of the basis set on excitation energies and state order was also investigated in this work; despite the differences in excitation energies, the state orders remain the same for all the methods independently of the basis set used (see Supporting Information). ADC(s)-x is not included in this discussion since it gives an overall unbalanced description of the excited states and an underestimation of the excitation energies (see Supporting Information) [Citation44]. One observes the typical blueshift of all four lowest states to higher excitation energies when the fraction of Hartree-Fock (HF) exchange is increased from zero (BLYP) to 20% (B3LYP), and finally to 50% (BHLYP). The state order remains the same for the functionals with HF fraction higher than zero and it is similar to the state order in ADC(2)-s and EOM-CCSD (see Table ). These suggest that tetracene can be satisfactorily described by TD-DFT because of its closed-shell configuration, being also a planar molecule not prone to intramolecular charge transfer. In addition, increasing the size of the basis set by adding more diffuse basis functions has negligible effect on excitation energy and state order, suggesting these state being local excited states [Citation31].
Table 1. Excitation energies (in eV) and oscillator strengths (in parentheses) for the four lowest singlet excited states of a tetracene molecule calculated at different levels of theory with ANO-S-VDZP as basis set.
From Table , ADC(2)-s/ANO-S-VDZP and EOM-CCSD/ANO-S-VDZP, have a very close agreement to the gas-phase experimental value for by 0.04 and 0.12 eV, respectively. However, also for
the excitation energy is overestimated by 0.53 eV (ADC(2)-s) and 0.61 eV (EOM-CCSD) when compared against the value measured in the crystal structure of 2.39 eV, [Citation50] and this can be attributed to the fact that no environmental effects were considered in our calculations. The
excitation energies calculated with ADC(2)-s and EOM-CCSD are also in line with a previously reported excitation energy calculated at the CASPT2/ANO-S-VDZP level of theory (2.87 eV [Citation48]) by about 0.05 and 0.13 eV, respectively. In the case of the remaining states where to the best of our knowledge no experimental values are available, ADC(2)-s and EOM-CCSD have a close agreement for
(0.01 eV of difference) but they deviate for the higher
and
states where the differences are approximately 0.4 eV. In the following, the method selection is restricted to the spectroscopically relevant states
and
(usually termed as
and
), whereas
and
will be kept and discussed to show the applicability of the exciton descriptors.
Comparing the performance of TD-DFT and ADC(2)-s wave function-based methods, i.e. ADC(2)-s and EOM-CCSD in Table , it is apparent that functionals containing larger amount of HF exchange (>50%), i.e. BHLYP, ωB97X-D, and CAM-B3LYP, perform in a close agreement (difference of eV) with ADC(2)-s and EOM-CCSD. A similar trend is observed for
, with slightly higher deviation (difference of
eV). Earlier studies on tetracene [Citation51, Citation52] and other large π-conjugated systems [Citation31, Citation33] have revealed that CAM-B3LYP is a suitable functional when describing excitation energies [Citation51] and exciton descriptors [Citation31, Citation33, Citation52]. Following these conclusions, the exciton descriptors (Ω,
,
,
,
and
) for the four lowest singlet excited states were then calculated with the long-range corrected functionals ωB97X-D and CAM-B3LYP and compared with the wave function-based methods, i.e. ADC(2)-s and EOM-CCSD; Table collects these results.
Table 2. Exciton descriptors of the four lowest singlet excited states of the tetracene molecule calculated at different levels of theory with ANO-S-VDZP as basis set.
The difference in Ω values between TD-DFT and ADC(2)-s and EOM-CCSD in Table comes from the fact that TD-DFT (within the adiabatic approximation) does not consider double and/or higher excitations, and therefore the transitions are predicted as pure single excitation character for all functionals. In contrast, values lower than one obtained with ADC(2)-s () and EOM-CCSD (
) can be attributed either to orbital relaxation effects or to a partial double excitation character of the state. For our purpose, these effects are not explored but a further discussion on how to identify and study them can be found in Ref. [Citation24].
The values of which are zero for all states independently of the method of choice, indicate that these states do not have pronounce CT character and can be safely labelled as local excitations. However,
values (
calculated for both TD-DFT and the wave function-based methods suggest that these states are not pure local excitations and contain some CT character. This will be further discussed in relation to the corresponding electron-hole correlation plots presented later in this section. It is worth to point out that both
and
depend on the fragmentation scheme used.
values in Table are calculated with the fragmentation scheme in Figure . Calculating
and creation of electron-hole correlation plots for EOM-CCSD is not currently supported in TheoDORE.
An overall inspection of the ,
,
and
descriptors, in Table reveals that the calculated values for the states of interest in this work, i.e.
and
, are very similar for all employed levels of theory. For the higher lying states
and
states, slight deviation can be observed. If one focuses on
values at ADC(2)-s and EOM-CCSD level of theories for
and
, it becomes evident that they show a different state order. A possible explanation for that is the small energy gap between the two states (see Table ). Therefore, the analysis and interpretation of these states is not straightforward.
Local excitation character for the and
states is supported by the fact that
(less than 0.1 Å) at all level of theories applied here; similar hole and electron sizes indicate valence states [Citation31]. Interestingly,
and
calculated using ωB97X-D and CAM-B3LYP functionals are very similar to the corresponding values calculated at ADC(2)-s and EOM-CCSD level of theories. The
magnitudes for the
and
states are similar between TD-DFT and the wave function-based methods, in the case of
this is in the order of
and for
between the 0.180–0.262 range. Moreover, all of them are positive values (for
and
) which indicates an attractive correlation between the electron and the hole.
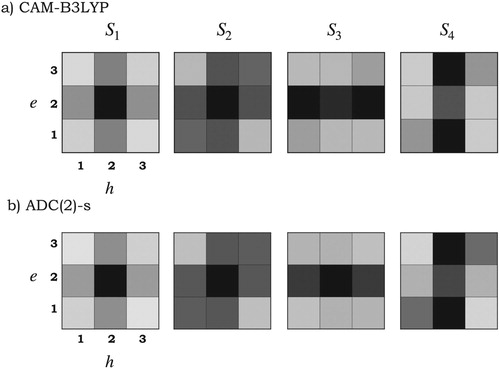
In Figure the electron-hole correlation plots of the tetracene molecule calculated with CAM-B3LYP and ADC(2)-s for the fragmentation scheme depicted in Figure are shown. For the state, the exciton is slightly delocalised over the whole system with the main contribution in the central fragment (fragment 2 in Figure ). Some CT occurs between neighbouring fragments but in a lesser extent (grey areas in the off-diagonal elements), as predicted from the
values in Table . In
we observe that the exciton is also delocalised across the molecule as in
but (1) the contribution from the central fragment is reduced, (2) the local excitation character in the extremes is enhanced, and (3) the CT between nearest neighbours is still present and slightly enhanced. The behaviour changes in higher excited states, e.g.
is predicted to be a local excitation with CT occurring from the benzene rings in the extremes towards the central fragment (from fragments 1 and 3 to 2).
is also a local exciton but the CT occurs from the central fragment towards the benzene rings (from fragment 2 to 1 and 3). The electron-hole correlation plots calculated with CAM-B3LYP are very similar to those obtained with ADC(2)-s (see Figure ), which corroborates the Coulomb-attenuated functional as a suitable functional for studying larger systems, namely tetracene pairs presented in Section 4.2.
Figure 3. Electron-hole correlation plots calculated at the (a) CAM-B3LYP/ANO-S-VDZP and (b) ADC(2)-s/ANO-S-VDZP level of theory for the four lowest singlet excited states of the tetracene molecule. e and h denote the electron and hole coordinates, respectively. Numbers represent the units in the fragmentation scheme depicted in Figure .
NTO transitions, weights and character of the states are shown in Table and the state-averaged NTOs, obtained with CAM-B3LYP, involved in the transitions are depicted in Figure . In this discussion, the NTOs corresponding to the hole are represented as and the electron (or particle) as
with i being the corresponding ith orbital. As discussed at the end of Section 2.1, exciton descriptors allow us to go beyond the common approach of simply analysing orbitals. Particularly when the orbitals of interest look very similar to each other and no precise conclusions can be done from them, which is the case for the NTOs of tetracene in Figure . The delocalization across the molecule is similar for the six orbitals involved in the transitions of the four lowest singlet states. Nevertheless, the character of the states can be assigned based in the NTOs. For instance,
has a
character, with the transition occurring from
to
with a weight of 0.94, which means that 94% of the
state is described by this NTO transition.
is composed by two NTO transitions of almost equal weight (0.51 and 0.46) and it corresponds to an overall
character. For
, the transition goes from
to
with a high weight of 0.95 and it corresponds to a
character. Similar results can be seen for the
state. The results for CAM-B3LYP are similar to those obtained with ADC(2)-s and EOM-CCSD (see Table ). The NTOs for ADC(2)-s and EOM-CCSD are provided in the Supporting Information.
Figure 4. (a) Hole and (b) particle state-averaged natural transition orbitals involved in the transitions of the four lowest excited states calculated at the CAM-B3LYP/ANO-S-VDZP level of theory. Isovalue=0.05.
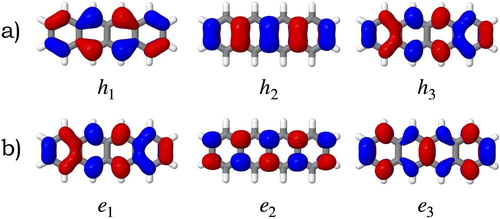
Table 3. NTO transitions, weights and character of the four lowest singlet excited states of the tetracene molecule calculated at CAM-B3LYP, ADC(2)-s, and EOM-CCSD level of theories using ANO-S-VDPZ as basis set.
The interpretation dependency on the fragmentation scheme has been previously discussed [Citation34]. To explore this influence, the tetracene molecule was fragmented asymmetrically in two, four and five units and the electron-hole correlation plots are shown in the Supporting Information. The plots show patterns that are difficult to interpret and relate to the NTO analysis. Previously another scheme with five fragments in tetracene was reported [Citation32]. Symmetric fragmentation is suggested to be the most suitable choice when possible, as it is done in this work, and if the units are not chemically equivalent the results should be analysed with care [Citation32].
From the discussion so far, the ωB97X-D and CAM-B3LYP seem to be the most suitable functionals for studying the excited states of tetracene. Based on the exciton descriptors, both functionals seem to give similar results, however CAM-B3LYP excitation energies are closer to the ADC(2)-s, EOM-CCSD and CASPT2(12,12) values (see Table ). CAM-B3LYP has been also reported previously as a suitable functional for the study of tetracene [Citation32, Citation38, Citation51, Citation52]. For studying clusters of N tetracene molecules, where CT between the monomers can occur, ωB97X-D and CAM-B3LYP would be proper choices. Both are long range corrected functionals, and they have showed a better performance when describing CT excitations [Citation53, Citation54].
4.2. Tetracene pairs in the crystal structure
The trimeric structure highlighted in Figure was taken from an optimised crystal structure (see Supporting Information), where three different repeating pairs of tetracene molecules were identified and named as AB, AC and BC for the upcoming discussion. Electronic couplings between the photoexcited state and the
state were calculated using the NOCI approach as described in Section 2.2 for the three pairs of molecules. Results in Table indicate that the electronic coupling is enhanced when the CT states are allowed to mix with the
and
states, which supports the superexchange-mediated mechanism as reported previously for tetracene [Citation22, Citation55]. AB pair has the highest electronic coupling (
meV), BC has the lowest (
meV) and AC value lies between the other two (
meV). As it was discussed in Section 2.2, the SF probability is proportional to the electronic coupling according to the Fermi's golden rule [Citation39], suggesting that AB pair shows the highest SF probability and BC pair the lowest. Here we present an excited state analysis to provide detailed insights into the nature of the excited states for the these pairs, having different relative orientations and electronic couplings, to identify whether there is any relation between the calculated electronic coupling and the character of the involved excites states.
Figure 5. Trimer arrangement highlighted within the tetracene optimised crystal structure, where three pairs of molecules (AB, AC and BC) are identified.
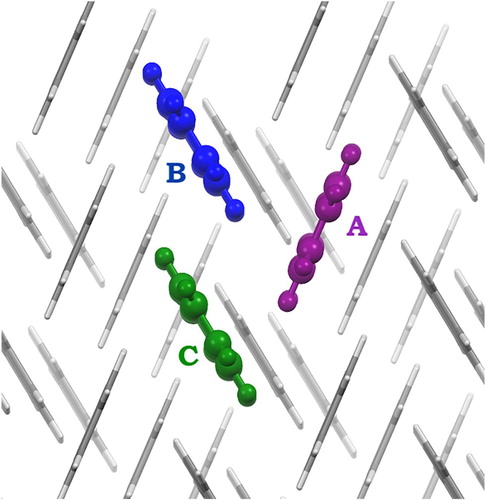
Table 4. Electronic couplings (in meV) between the (diabatic) states  / for the tetracene pairs AB, AC and BC. In the primed wave functions the CT states were allowed to mix with the and states.
/ for the tetracene pairs AB, AC and BC. In the primed wave functions the CT states were allowed to mix with the and states.
As for the monomer, we observe for all pairs a smooth and systematic blueshift in the excitation energies when increasing the amount of HF exchange (see Supporting Information). No artificial low-lying CT states were observed for the three pairs which means that TD-DFT can be used safely. The exciton descriptors computed at the CAM-B3LYP/ANO-S-VDZP level of theory for the AB, AC and BC pairs are presented in Table . Exciton descriptors calculated with ωB97X-D are shown in the Supporting Information. The fragmentation scheme used to calculate in the tetracene pairs is shown in Figure . We observed that for all states in the three pairs
(omitted in Table ) which indicates a pure single excitation character and arise from TD-DFT considering only single excitation and no double or higher excitations. Interested reader is referred to Ref. [Citation24] for how to identify and study the effect of higher excitation.
Figure 6. Labelled fragmentation scheme and electron-hole correlation plots for the three tetracene pairs of molecules AB, AC and BC calculated at the CAM-B3LYP/ANO-S-VDZP level of theory. e and h represent the electron and hole coordinates, respectively.
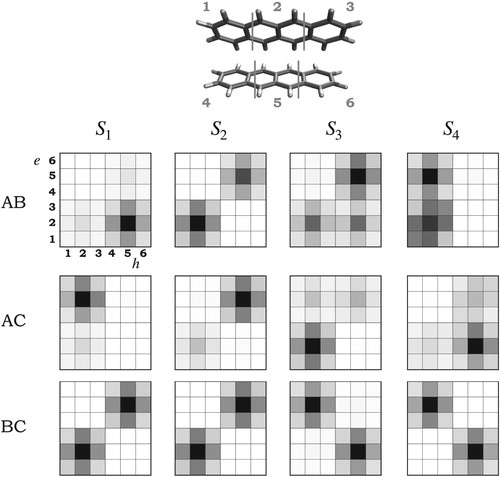
Table 5. Exciton descriptors for the tetracene pairs (AB, AC and BC) calculated at the CAM-B3LYP/ANO-S-VDZP level of theory.
Differences between the pairs with large electronic coupling (AB and AC) and the pair with zero electronic coupling (BC) start to be evident when analysing the remaining exciton descriptors in Table . For the AB pair, shows a CT character since
Å and
. The fact that
indicates that the state has no pure CT character, but rather is mixed with the local excited states which lie very close in energy.
for
matches with the distance of separation between the tetracene molecules in the AB pair (3.819 Å) suggesting that the electron and hole are located each in a different tetracene molecule. This opposite localisation of the hole and electron in the pair arrangement is also reflected in the large
(6.205 Å). The low positive value of
can be also interpreted as a loosely Coulombic attraction between the electron and hole which are located at a large distance of separation. Interestingly, the sizes of the electron and hole are the same (
). For
the interpretation is not as straightforward as for
; firstly,
indicates a CT state substantially mixed with the local excitations, which leads to a lowering of the
to 1.994 Å and
to 5.355 Å and in increasing in the electron-hole correlation of
. In contrast,
is a local excited state with
where some CT is occurring; these CT is reflected in the almost zero value of
. Both,
and low
value (4.652 Å) indicate a local excitation in a single tetracene molecule. The fact that electron and hole are located in the same molecule is reflected in a larger positive
(0.368) understood as a stronger Coulombic attraction. For
, the state is considered as a local excitation (
) in one of the tetracene molecules where the CT mixing is enhanced in comparison with the
state which leads to a higher
(1.044 Å) and a lower
(0.209).
A very similar interpretation as in AB can be made for AC pair, where basically the same behaviour can be observed for the four states with and
as predominantly CT states (
Å), while
and
are local excitons (
Å) with some CT character. Larger
values indicate CT states and lower
local excited states. Although positive
values indicate joint excitons in the four states, the lower values for the CT states when compared to the local excitations reveal that the spatial correlation is less pronounced. This indicates that the electron and hole in the CT states are loosely correlated due to their long exciton length.
For the BC pair, with zero electronic coupling, the state order is completely different than for AC and AB. All states have which suggest local excitation character, but just
and
can be identified as excitons due to their lower
values (
) and the relative high positive
values (
). The larger
values for the local excitations in pair BC suggest a stronger Coulombic attraction of the hole and electron, than in the AB and AC pairs.
and
states can be identified as CR states, due to their
values close to one and the negative
values [Citation36]. The
for both states can be explained due to the parallel symmetry of the pair arrangement. The difference between the states is also reflected in
where the CR states have higher values (> 7 Å) than the excitonic states (< 4.7 Å).
The electron-hole correlation plots of the three pairs are depicted in Figure , and the state-averaged NTO pairs involved in the transitions are shown in Figure . These tools give a more detailed insight on the excited states of the tetracene pairs. For AB and AC pairs, the CT states and
correspond to transitions between fragments 2 and 5 (as labelled in Figure ) which corresponds to the middle part of the tetracene molecules. The CT character of the states is supported by the NTO analysis, e.g.
is noticeably more localised in the middle fragment of one of the molecules whereas
is localised on the central part of the opposite molecule. For the
in AB pair, a small local excitation character is expected.
is composed by two excitations, in both (according to the plots) the hole would be localised in fragment 2 whereas the electron would not be only on fragment 5 but also in fragment 2. A similar analysis holds for
in AC. The localised
and
excited states, correspond to
transitions. Remarkably, the electron-hole correlation plots suggest that the exciton is localised in one of the molecules in the AB and AC pairs, which is line with previously reported observations [Citation13, Citation20]. Additionally, the CT between neighbouring chromophores inferred by the exciton descriptors is confirmed here by the grey off-diagonal elements.
Figure 7. State-averaged natural transition orbitals involved in the five lowest singlet excited states for the pairs AB, AC and BC calculated at the CAM-B3LYP/ANO-S-VDZP level of theory. Isovalues = 0.05.
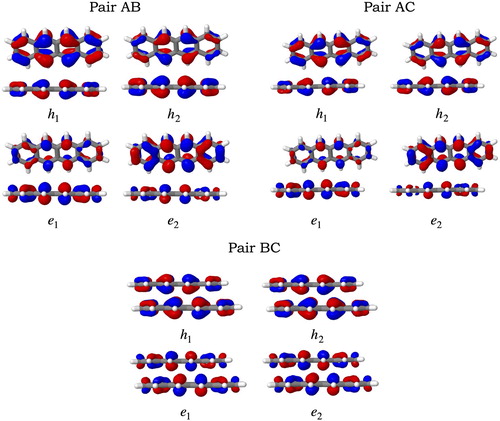
For the BC pair, the and
states correspond both to excitations delocalised in both monomers equally, which differs from the local exciton in the AB and AC pairs. Small CT contributions are predicted for these states. Both states are composed of two transitions (both of
character) as stated in Table , where the NTOs reveal that the orbitals are delocalised in the middle fragments of both molecules.
The exciton descriptors confirm that the AB and AC pairs, with non-zero electronic couplings, have low-lying CT states close in energy to the local excited states. values and the electron-hole correlation plots in Figure show that there is mixing between these states for AB and AC pairs. This supports the CT-mediated superexchange mechanism inferred by the enhancement of electronic couplings when CT states are mixed with the
and
states in these two pairs. Results obtained with the ωB97X-D functional predict mostly pure LE excitations, which does not reflect the enhancement of the electronic coupling via CT mixing. In contrast in the BC pair, with zero electronic coupling, high-lying CR states are predicted, which could not be inferred from the electronic coupling study. Based on these observations, the difference in electronic couplings might be then attributed to the presence and mixing in of CT states, while CR states were observed in the pair with zero electronic coupling. Whether CR states play any role in the overall SF process need to be clarified and studied further. More important for SF process, that is the focus of this work, is the interesting trends observed for electron-hole correlation coefficient,
; for the AB and AC pairs the local excitations (
and
) are expected to have a weaker Coulombic attraction (
and 0.209 for AB,
and 0.246 for AC) in comparison with the local excitations (
and
) in pair BC (
). We speculate that the excited electron could be more easily transferred to a neighbouring molecule in pairs AB and AC, due to the weaker Coulombic attraction. This might facilitate the CT-mediated superexchange mechanism for the formation of the
state from the photoexcited molecule (see Figure ).
5. Conclusions
In this study we have performed an excited state analysis on tetracene molecule, in its monomeric, dimeric and trimeric forms, to identify the role of CT states in the underlying mechanism through which the SF process occurs. Our study revealed that CAM-B3LYP functional shows the best overall agreement in terms of excitation energies and exciton descriptors, based on the one-particle transition density matrix, when compared with the wave function-based methods ADC(2)-s and EOM-CCSD, and therefore used as the most suitable and computationally affordable methodology to further study tetracene pairs. The dependency of the fragmentation scheme in the interpretation of results was explored; a symmetrical fragmentation for acenes is suggested for further studies when possible. In the particular case of tetracene, a three-units fragmentation scheme was presented and explored. Exciton descriptors, electron-hole correlation plots, and NTOs were computed for three pairs of tetracene molecules, with different relative orientations and electronic couplings, and then used to assign the character of their excited states. Our results show that the exciton descriptors can differentiate between CT and CR states. The presence of low-lying CT states, mixed with the local excitations, are found in pairs with non-zero electronic coupling (AB and AC) which indicates that the conversion to the states is enhanced and most probably occur through a CT-mediated superexchange mechanism. Relatively high-lying CR states are observed in the pair with zero electronic coupling (BC). Moreover, we have found that excitons in AB and AC pairs have a weaker Coulombic attraction (indicated by their lower electron-hole correlation coefficient) than the exciton in BC pair, and therefore they might easily undergo through SF conversion to the
state. In summary, our work has shown the applicability of the excited state analysis to characterize the excited states of the tetracene pairs, with different relative orientation and electronic coupling, and to differentiate the role that CT and CR states play in the underlying mechanism of the SF process. The analysis can be extended to study the influence of vibrational modes and environmental effects in the electronic couplings and the excited states of the tetracene pairs or other potential molecules exhibiting SF of comparable size and complexity.
Acknowledgments
This research used resources of the Oak Ridge Leadership Computing Facility located at Oak Ridge National Laboratory, which was supported by the Office of Science of the Department of Energy under Contract No. DEAC05-00OR22725. This work is part of Innovational Research Incentives Scheme Vidi 2017 with project number 016.Vidi.189.044, which is (partly) financed by the Dutch Research Council (NWO).
Disclosure statement
No potential conflict of interest was reported by the author(s).
ORCID
Luis Enrique Aguilar Suarez http://orcid.org/0000-0001-6983-5151
Maximilian F. S. J. Menger http://orcid.org/0000-0003-1442-9601
Shirin Faraji http://orcid.org/0000-0002-6421-4599
Additional information
Funding
References
- C.D. Thomas, A. Cameron, R.E. Green, M. Bakkenes, L.J. Beaumont, Y.C. Collingham, B.F.N. Erasmus, M.F. de Siqueira, A. Grainger, L. Hannah, L. Hughes, B. Huntley, A.S. van Jaarsveld, G.F. Midgley, L. Miles, M.A. Ortega-Huerta, A. Townsend Peterson, O.L. Phillips and S.E. Williams, Nature427 (6970), 145–148 (2004). doi: 10.1038/nature02121
- C. Li, F. Wang and C. Yu, Energy Environ. Sci. 4, 100–113 (2011). doi: 10.1039/C0EE00162G
- J.L. Bredas, J.E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res. 42 (11), 1691–1699 (2009). doi: 10.1021/ar900099h
- M.B. Smith and J. Michl, Chem. Rev. 110 (11), 6891–6936 (2010). doi: 10.1021/cr1002613
- J. Lee, P. Jadhav, P.D. Reusswig, S.R. Yost, N.J. Thompson, D.N. Congreve, E. Hontz, T. van Voorhis and M.A. Baldo, Acc. Chem. Res. 46 (6), 1300–1311 (2013). doi: 10.1021/ar300288e
- A. Rao and R.H. Friend, Nat. Mater. 2 (11), 17063 (2017). doi: 10.1038/natrevmats.2017.63
- A.J. Nozik, R.J. Ellingson, O.I. Micic, J.L. Blackburn, P. Yu, J.E. Murphy, M.C. Beard and G. Rumbles, in Proceedings of the 27th DOE Solar Photochemistry Research Conference (Warrenton, Virginia), pp. 63–66.
- M.C. Hanna and A.J. Nozik, J. Appl. Phys. 100, 074510 (2006). doi: 10.1063/1.2356795
- M.H. Futscher, A. Rao and B. Ehrler, ACS Energy Lett. 3 (10), 2587–2592 (2018). doi: 10.1021/acsenergylett.8b01322
- B. Ehrler, M.W.B. Wilson, A. Rao, R.H. Friend and N.C. Greenham, Nano Lett. 12 (2), 1053–1057 (2012). doi: 10.1021/nl204297u
- P.D. Reusswig, D.N. Congreve, N.J. Thompson and M.A. Baldo, Appl. Phys. Lett. 101, 113304 (2012). doi: 10.1063/1.4752445
- I. Paci, J.C. Johnson, X. Chen, G. Rana, D. Popovia, D.E. David, A.J. Nozik, M.A. Ratner and J. Michl, J. Am. Chem. Soc. 128 (51), 16546–16553 (2006). doi: 10.1021/ja063980h
- L.E.A. Suarez, R.K. Kathir, E. Siagri, R.W.A. Havenith and S. Faraji, Adv. Quant. Chem. 79, 263–287 (2019). doi: 10.1016/bs.aiq.2019.05.004
- D. Casanova, Chem. Rev. 118 (15), 7164–7207 (2018). doi: 10.1021/acs.chemrev.7b00601
- J.M. Herbert, X. Zhang, A.F. Morrison and J. Liu, Acc. Chem. Res. 49 (5), 931–941 (2016). doi: 10.1021/acs.accounts.6b00047
- N. Elfers, I. Lyskov, J.D. Spiegel and C.M. Marian, J. Phys. Chem. C 120 (26), 13901–13910 (2016). doi: 10.1021/acs.jpcc.6b02263
- P.M. Zimmerman, F. Bell, D. Casanova and M. Head-Gordon, J. Am. Chem. Soc. 133 (49), 19944–19952 (2011). doi: 10.1021/ja208431r
- P.M. Zimmerman, C.B. Musgrave and M. Head-Gordon, Acc. Chem. Res. 46 (6), 1339–1347 (2013). doi: 10.1021/ar3001734
- A.F. Morrison and J.M. Herbert, J. Chem. Phys 146, 224110 (2017). doi: 10.1063/1.4985607
- R.W.A. Havenith, H.D. de Gier and R. Broer, Mol. Phys. 110 (1920), 2445–2454 (2012). doi: 10.1080/00268976.2012.695810
- M. Wibowo, R. Broer and R.W.A. Havenith, Comput. Theor. Chem. 1116, 190–194 (2017). doi: 10.1016/j.comptc.2017.03.013
- S.R. Yost, J. Lee, M.W. Wilson, T. Wu, D.P. McMahon, R.R. Parkhurst, N.J. Thompson, D.N. Congreve, A. Rao, K. Johnson and M.Y. Sfeir, Nature Chem. 6, 492–497 (2014). doi: 10.1038/nchem.1945
- F. Plasser, M. Wormit and A. Dreuw, J. Chem. Phys. 141, 024106 (2014).
- F. Plasser, S.A. Bappler, M. Wormit and A. Dreuw, J. Chem. Phys. 141, 024107 (2014).
- N. Geacintov, M. Pope and F. Vogel, Phys. Rev. Lett. 22 (12), 593–596 (1969). doi: 10.1103/PhysRevLett.22.593
- R.P. Groff, P. Avakian and R.E. Merrifield, Phys. Rev. B 1 (2), 815–817 (1970). doi: 10.1103/PhysRevB.1.815
- J.J. Burdett, A.M. Müller, D. Gosztola and C.J. Bardeen, J. Chem. Phys. 133, 144506 (2010). doi: 10.1063/1.3495764
- A.F. Morrison and J.M. Herbert, Phys. Chem. Lett. 8 (7), 1442–1448 (2017). doi: 10.1021/acs.jpclett.7b00230
- X. Feng, A.V. Luzanov and A.I. Krylov, J. Phys. Chem. Lett. 4 (22), 3845–3852 (2013). doi: 10.1021/jz402122m
- C Ullrich, Time-Dependent Density-Functional Theory. Concepts and Applications, Oxford Graduate Texts (Oxford University Press, Great Britain, 2012).
- S.A. Mewes, F. Plasser, A.I. Krylov and A. Dreuw, J. Chem. Theory Comput. 14 (2), 710–725 (2018). doi: 10.1021/acs.jctc.7b01145
- S.A. Bäppler, F. Plasser, M. Wormit and A. Dreuw, Phys. Rev. A 90, 052521 (2014). doi: 10.1103/PhysRevA.90.052521
- S.A. Mewes and A. Dreuw, Phys. Chem. Chem. Phys. 21, 2843–2856 (2019). doi: 10.1039/C8CP07191H
- S.A. Mewes and J.M. Mewes, Phys. Chem. Chem. Phys. 18, 2548–2563 (2016). doi: 10.1039/C5CP07077E
- F. Plasser, ChemRxiv, 10.26434/chemrxiv.11395314.v1 (2019).
- F. Plasser, B. Thomitzni, S.A. Bäppler, J. Wenzel, D.R. Rehn, M. Wormit and A. Dreuw, J. Comp. Chem. 36, 1609–1620 (2015). doi: 10.1002/jcc.23975
- P. Kimber and F. Plasser, Phys. Chem. Chem. Phys. 22, 6058–6080 (2020). doi: 10.1039/D0CP00369G
- S.A. Mewes, F. Plasser and A. Dreuw, J. Phys. Chem. Lett. 8 (6), 1205–1210 (2017). doi: 10.1021/acs.jpclett.7b00157
- C.H. Yang and C.P. Hsu, J. Phys. Chem. Lett. 6 (10), 1925–1929 (2015). doi: 10.1021/acs.jpclett.5b00437
- E.A. Buchanan, Z. Havlas and J. Michl, Adv. Quant. Chem. 75, 175–227 (2017). doi: 10.1016/bs.aiq.2017.03.005
- T.P. Straatsma, R. Broer, S. Faraji and R.W.A. Havenith, Annu. Rep. Comput. Chem. 14, 77–91 (2018). doi: 10.1016/bs.arcc.2018.06.003
- Z.Q. You and C.P. Hsu, Int. J. Quantum Chem. 114, 102–115 (2014). doi: 10.1002/qua.24528
- S. Grimme, J. Comp. Chem. 27, 1787 (2006). doi: 10.1002/jcc.20495
- A. Dreuw and M. Wormit, Comput. Mol. Sci. 5, 82–95 (2015). doi: 10.1002/wcms.1206
- Y. Shao, Z. Gan, E. Epifanovsky, A.T. Gilbert, M. Wormit, J. Kussmann, A.W. Lange, A. Behn, J. Deng, X. Feng and D. Ghosh, Mol. Phys. 113, 184–215 (2015). doi: 10.1080/00268976.2014.952696
- F. Plasser, TheoDORE: a package for theoretical density, orbital relaxation and exciton analysis. <http://theodore-qc.sourceforge.net>.
- M.F. Guest, I.J. Bush, H. van Dam, P. Sherwood, J. Thomas, J. van Lenthe, R.W.A. Havenith and J. Kendrick, Mol. Phys. 103 (6–8), 719–747 (2005). doi: 10.1080/00268970512331340592
- F. Plasser, S.A. Mewes, A. Dreuw and L. González, J. Chem. Theory Comput. 13 (11), 5343–5353 (2017). doi: 10.1021/acs.jctc.7b00718
- S. Grimme and M. Parac, Chem. Phys. Chem. 4, 292–295 (2003). doi: 10.1002/cphc.200390047
- H.B. Klevens and J.R. Platt, J. Chem. Phys. 17 (5), 470–481 (1949). doi: 10.1063/1.1747291
- M.J.G. Peach, P. Benfield, T. Helgaker and D.J. Tozer, J. Chem. Phys. 128, 044118 (2008). doi: 10.1063/1.2831900
- S.A. Mewes, F. Plasser and A. Dreuw, J. Chem. Phys. 143, 171101 (2015). doi: 10.1063/1.4935178
- T. Yanai, D.P. Tew and N.C. Handy, Chem. Phys. Lett. 393, 51–57 (2004). doi: 10.1016/j.cplett.2004.06.011
- J.D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys. 10, 6615–6620 (2008). doi: 10.1039/b810189b
- H. Yamagata, J. Norton, E. Hontz, Y. Olivier, D. Beljonne, J.L. Bredas, R. Silbey and F. Spano, J. Chem. Phys. 134, 204703 (2011). doi: 10.1063/1.3590871