

Abstract
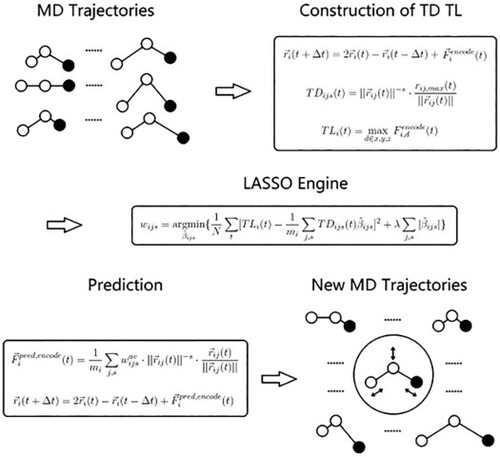
Ab initio molecular dynamics (AIMD) has become a popular simulation technique but long simulation times are often hampered due to its high computational effort. Alternatively, classical molecular dynamics (MD) based on force fields may be used, which, however, has certain shortcomings compared to AIMD. In order to alleviate that situation, a trajectory-based machine learning (TrajML) approach is introduced for the construction of force fields by learning from AIMD trajectories. Only nuclear trajectories are required, which can be obtained by other methods beyond AIMD as well. We developed an easy-to-use MD machine learning package (TrajML MD) for instant modelling of the force field and system-focussed prediction of molecular configurations for MD trajectories. It consumes similar computational resources as classical MD but can simulate complex systems with a higher accuracy due to the targeted learning on the system of interest.
GRAPHICAL ABSTRACT
Acknowledgements
Funding by the University of Zurich and the Swiss National Science Foundation (grant no: PP00P2_170667) is gratefully acknowledged. We thank the Swiss National Supercomputing Center for computing resources (project ID: s745 and s788).
Disclosure statement
No potential conflict of interest was reported by the author(s).