
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A quasi-relativistic (two-component) zeroth-order regular approximation model potential approach is employed to calculate nuclear electric quadrupole coupling tensors of the chiral polyhalomethane derivatives CHBrClF and CHClFI. Each of these two chiral compounds contains two nuclei with electric quadrupole moments, which poses special challenges for an analysis of high-resolution rovibrational spectra and, thus, renders predictions of electric field gradients and resulting nuclear electric quadrupole coupling tensors in such molecules particularly valuable. The theoretical approach is studied with respect to the influence of relativistic effects, by comparison to scalar-relativistic and non-relativistic calculations, and electron correlation effects, which are included within different flavours of density functional theory. The quality of the calculations is assessed by comparison to results from experiments reported previously by Bauder et al. and Soulard et al., respectively. It is shown that the local density approximation exchange-correlation functional gives results that agree with experiment within 12 % for all nuclear quadrupole coupling tensor components of 79Br and 127I. This benchmark test of the quasi-relativistic approach indicates its applicability to support upcoming microwave experiments with chiral heavy-element containing main group compounds.
GRAPHICAL ABSTRACT
1. Introduction
Nuclear electric quadrupole moments (NEQMs) are a window into nuclear structure [Citation1]. Presently, the most accurate method for determining NEQMs relies on the fruitful interplay between atomic or molecular precision spectroscopy and electronic structure theory [Citation2]. In atomic and molecular spectroscopy NEQM coupling tensors are determined, which are a product of the NEQM and the electric field gradient (EFG) tensor probed at the position of the nucleus. The latter is not directly accessible by experiment, but can be calculated with electronic structure methods. Once the NEQM of a nucleus is determined, the measurement of NEQM coupling tensors in high-resolution rovibrational spectra of molecules provides important information in particular on the electronic density distribution in the vicinity of the nuclei. Due to this property, relativistic effects can play a major role in the calculation of EFGs of heavy element containing compounds [Citation3,Citation4]. As several atomic and molecular properties of current fundamental interest, such as a possible permanent electric dipole moment in molecules and atoms caused by parity- and time-reversal violating electric dipole moments of the nucleons, nuclear Schiff moments, nuclear magnetic quadrupole moments or an electric dipole moment of the electron, depend on the behaviour of the electronic wavefunction near the nucleus and benefit from relativistic enhancement effects [Citation5–7], NEQM coupling tensors can serve as tests for the accuracy of theoretical approaches for the description of symmetry violating effects in molecular systems.
Whereas for an accurate prediction of NEQM coupling tensors in molecules with light nuclei a perturbative description of relativistic effects gives excellent results (see for instance, the direct perturbation theory studies of Stopkowicz and Gauss in Refs. [Citation8,Citation9]), in heavier compounds relativistic effects are preferentially included self-consistently. Many different relativistic approaches, ranging from four-component Dirac–Hatree–Fock [Citation3,Citation4,Citation10] and coupled cluster methods [Citation11–15] to two- and one-component Douglas–Kroll–Hess (DKH) and zeroth-order regular approximation (ZORA) approaches [Citation16–21] have been considered. It was shown in some of these previous studies that ZORA performs well for the description of these relativistic effects in heavy elemental compounds [Citation16,Citation18,Citation19,Citation21] as the EFG pronouncedly depends on the behaviour of the valence electrons near the nuclei, whereas contribution from the core orbitals themselves remain usually below 15 % [Citation18].
Jürgen Gauss and colleagues have performed combined experimental and theoretical high-level studies of NEQM coupling tensors in rotational spectra of the methane derivatives CHBrF [Citation22,Citation23] and CH
FI [Citation24,Citation25], which feature each a single quadrupolar halogen nucleus. They were also able to extract NEQM coupling tensors of rare isotopes such as deuterium in the corresponding deuterated derivatives.
In high-resolution rotational spectroscopy of chiral polyhalogenated molecules, additional challenges have to be overcome as different halogen nuclei contribute to the spectral splittings in experiment [Citation26,Citation27]. Furthermore, the full NEQM coupling tensors needs to be determined. This renders quantum chemical calculations of NEQM couplings in chiral molecules particularly valuable.
In this paper, we demonstrate the applicability of model potential ZORA-DFT methods [Citation7,Citation28] for heavy-element containing chiral molecules that are of special interest for a measurement of molecular parity violation (see e.g. Ref [Citation29] for a recent review on molecular parity violation), i.e. the polyhalomethanes CHBrClF [Citation26,Citation30–37] and CHClFI [Citation33,Citation37,Citation38]. Both molecules were studied intensely by experiment giving a good reference to validate our methods for such chiral systems. Chiral compounds that contain even heavier nuclei will be in the focus of future research.
2. Methodology and computational details
The static interaction of a nuclear electric quadrupole moment (NEQM) described by the symmetric second rank tensor of nucleus A with the electron cloud of a molecule with
electrons and
nuclei is described in a four-component relativistic electronic structure framework by the Hamiltonian (see e.g. Ref. [Citation39]; we follow here the sign convention of Refs. [Citation26,Citation27]):
(1)
(1) with
denoting the negative of the EFG at nucleus A from electron i,
denoting the negative of the EFG at nucleus A from nucleus B and
being the number of electrons in the system.
The quadrupole moment tensor can be written in terms of the nuclear electric quadrupole moment as
, where
is a dimensionless symmetric second rank tensor depending on the nuclear spin. With this, we can also define an effective quadrupole coupling tensor
for the NEQM entering the effective spin Hamiltonian as
(2)
(2) with elements of the NEQM coupling tensor being (see e.g. Ref. [Citation40])
(3)
(3) Here
is the multi-electron wave function. The first of the terms in square brackets in Equation (Equation3
(3)
(3) ) is due to the contribution from the electrons in the system, whereas the second-term accounts for the electric field gradient caused by all other nuclei. For point-like particles, the negative of the EFG is in SI units
(4)
(4) with
for electrons and
for nuclei, with
being the nuclear charge number of nucleus B. As the point-nucleus potential has a pole at
there is an additional term to its derivative in the distributional sense proportional to
. This term does not contribute to the NEQC tensor and, thus, is not included in the present calculations. Here,
is the electric constant,
is the position vector and
is the Kronecker delta, with
and
denoting one of the Cartesian components x, y, z. Effects of the finite nuclear size on the form of the EFG operator are negligibly small for the molecules studied herein, but may become more pronounced for molecules that contain heavier elements as we have shown for the electric field at the nucleus in another context [Citation6].
Due to appearance of the identity matrix in the electronic expectation value in Equation (Equation3
(3)
(3) ), both the large and the small component densities of the relativistic electronic wavefunction contribute within a four-component formulation of the EFG operator to the electronic part of Equation (Equation4
(4)
(4) ). When the multi-electron wave function is approximated as a single determinant composed from
four-component molecular orbitals
with large (or upper) components
and small (or lower) components
, the contribution to the electronic part reads as
(5)
(5) with
being the occupation number of molecular orbital i and with 1 serving as a dummy number to indicate the single electron occupying the molecular orbital.
In the present paper, we follow our toolbox approach for the calculations of two-component properties within ZORA [Citation7]. For this purpose, we compute the ZORA Hartree–Fock (HF) or Kohn–Sham (KS) molecular orbitals by solving the ZORA equations self-consistently:
(6)
(6) where
is the model potential as proposed by van Wüllen [Citation28], which is employed in order to alleviate the gauge dependence of ZORA. and
is the effective potential within the complex generalised HF (cGHF) or KS (cGKS) approximation.
In this implementation, the large component is approximated as the ZORA wave function and terms involving the small component can be calculated by automatic generation of an approximate small component in the sense of ZORA:
(7)
(7) In the sense of our toolbox approach [Citation7], the electronic EFG operator can be represented by the four component density function
, a tensor operator
, a scalar operator
and no differential operator, where we have used the notation detailed in Ref. [Citation7]. Within this notation, contributions to
from the approximate ZORA two component number density functions
and
have to be evaluated. The contribution of the nuclear-nuclear repulsion was directly evaluated as written in Equation (Equation3
(3)
(3) ) and added subsequently to the electronic contribution to receive the NEQC tensor. For more details on our toolbox approach and the corresponding notation, we refer the reader to Ref. [Citation7].
In the following, we will refer to property calculations that include contributions from both and
and use the ZORA orbitals in renormalised form via
as (LL+SS) and to those which consider contributions from only
and do not use renormalised orbitals as (LL). The neglect of the contributions from
and from renormalisation is commonly referred to as the picture change error, which is given herein by the difference between LL+SS and LL results. It shall be noted that, for the EFG operator, corrections stemming from renormalisation remain essentially negligible as mainly valence orbitals contribute to the expectation value. One commonly accounts for nuclear electric quadrupole moments within first-order perturbation theory only, so that contributions from electric quadrupole moments of the various nuclei remain additive.
Quasi-relativistic two-component (2c) calculations are performed in this work within ZORA at the level of cGHF or cGKS with a modified version [Citation7,Citation41–46] of the quantum chemistry program package Turbomole [Citation47] to determine the unperturbed HF or KS molecular orbitals.
Structure parameters of CHBrClF and CHClFI, as well as electronic densities at the level of two-component ZORA (2cZORA) were employed as described in Ref. [Citation37]. Electronic densities were calculated on the level of one component ZORA-cGHF and ZORA-cGKS (1cZORA) and on the non-relativistic level of restricted HF (RHF) and restricted KS (RKS) with the same basis set employed in Ref. [Citation37]. Density functionals used in the present study are the local density approximation (LDA) [Citation48–50] as well as the Lee, Yang and Parr correlation functional (LYP) [Citation51] combined with a generalised gradient exchange functional by Becke (BLYP) [Citation52] or the hybrid Becke three parameter exchange functional (B3LYP)[Citation49,Citation53–55].
A ZORA-model potential as proposed by van Wüllen [Citation28] was employed with additional damping [Citation56].
In all self-consistent calculations of the unperturbed electron densities a finite nucleus was used, described by a normalised spherical Gaussian nuclear density distribution , where
and the root mean square radius
of nucleus A was used as suggested by Visscher and Dyall [Citation57]. The mass numbers A were chosen to correspond to the isotopes
H,
C,
F,
Cl,
Br,
I.
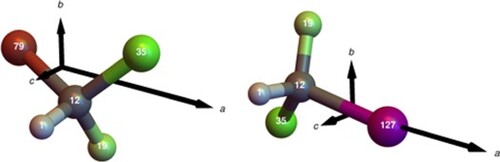
All electronic expectation values of electric field gradients were calculated by numerical integration on an ultra fine grid with our ZORA property toolbox approach described in Ref. [Citation7]. Resulting EFG tensors were multiplied by the corresponding value of the NEQM taken from Ref. [Citation2] and rotated into the principal axis system of the moment of inertia tensor following the same axis convention as used in Ref. [Citation26] and Ref. [Citation27], respectively (see Figure ).
Figure 1. Choice of principal axes in CHBrClF (left) and CHClFI (right) in centre of mass coordinates. Equilibrium structures are shown as optimised in Ref. [Citation37] at the level of CCSD(T) with small basis sets. The relative length chosen here for the principal axes corresponds to the relative size of the rotational constant. The a axis in CHClFI is scaled (shortened) by a factor of 2/3 relative to the b and c axes for a better representation. Atoms are labelled with mass numbers of used isotopes for and
.
![Figure 1. Choice of principal axes in CHBrClF (left) and CHClFI (right) in centre of mass coordinates. Equilibrium structures are shown as optimised in Ref. [Citation37] at the level of CCSD(T) with small basis sets. The relative length chosen here for the principal axes corresponds to the relative size of the rotational constant. The a axis in CHClFI is scaled (shortened) by a factor of 2/3 relative to the b and c axes for a better representation. Atoms are labelled with mass numbers of used isotopes for 12C1H79Br35Cl19F and 12C1H35Cl19F127I.](/cms/asset/348068ce-4fa5-47b4-9a5d-7a07140d6a3e/tmph_a_1797199_f0001_c.jpg)
3. Results
The rotational constants of the molecular structures obtained in Ref. [Citation37] at the level of CCSD(T) with a small basis set are MHz,
MHz and
MHz for CHBrClF and
MHz,
MHz and
MHz for CHClFI. Compared to the experimental rotational constants of the vibrational ground state reported in Refs. [Citation26,Citation27] these are off by 1 to 5 %. As EFGs can be rather sensitive to a change in molecular structure, this might slightly affect the accuracy of our results (see e.g. Ref. [Citation11]). In Tables and , the results for CHBrClF are listed and those of CHClFI can be found in Tables and .
3.1. CHBrClF
Relativistic effects are not important for and spin-orbit coupling effects are negligible for the NEQM coupling of both Cl and Br (
). Among the relativistic corrections, scalar-relativistic effects are most important (about
to
of the total relativistic effects for Br,
to
for Cl). The so-called picture change error has an influence of about 20 % on the total relativistic corrections, whereas spin-orbit coupling contributes only around 6 % to 8 % of the total relativistic effects for Br, and at most 5 % for Cl.
As there are only comparatively light nuclei (Z<50) in CHBrClF, electron correlation effects play a more prominent role than relativistic effects. Our calculations show that HF overestimates the absolute values of consistently by about 15 %.
In order to gauge the dependence on the molecular structure and the basis set and to allow for direct comparison to post-HF results obtained in Ref. [Citation58] on the second-order Møller–Plesset perturbation theory (MP2) level in the non-relativistic limit, we calculated also NEQM coupling tensors for the structure that was energy-minimised in Ref. [Citation58] at the level of MP2/6-311+G(2df,2pd). These additional calculations were performed at the HF level with the large even tempered basis set (see methodology and Ref. [Citation37]) (Hobi-G) and with the 6-311+G(2df,2pd) basis set (Hobi-B+G). Furthermore, with the structure of Ref. [Citation37] employed for all other calculations, the non-relativistic NEQM tensor was calculated at the level of HF/6-311+G(2df,2pd) (Hobi-B). These calculations show a weak dependence on the basis set ( for
and
) and a mild dependence on the molecular structure (
to
for the diagonal elements of
and
). The pure correlation effects, estimated as the difference between MP2 [Citation58] and HF results, are found to be on the order of
. This leads to deviations of our HF result (with basis and structure from Ref. [Citation37]) from the MP2 result of Ref. [Citation58] of up to
(aa component of
).
We tested density functionals of different flavour: hybrid, generalised gradient approximation (GGA) and LDA functionals. As to be expected, the largest deviation was found between LDA and HF, being on average of about 5 % (largest deviation is 11 % for the bb component of ). Electron correlation described by DFT is found to lower absolute values, which is in line with the correlation effects of MP2. Thus, our study shows that LDA gives the best results among the functionals considered here (up to 14 % deviation for
and up to 8 % deviation of
from experiment).
Assuming the structure dependence studied at the level of HF is the same for DFT methods, the NEQM coupling tensors may get even closer to experimental results if the molecular structure of Ref. [Citation58] is employed. For example, a correction of the 2cZORA(LL+SS)-LDA values by the difference between the HF/Hobi-G data and the HF results would yield deviations below from experiment for all components of
.
3.2. CHClFI
Whereas relativistic contributions do not play an important role for as in CHBrClF, relativistic effects become as expected very important for
and exceed electron correlation effects.
Scalar-relativistic effects are most important giving for of the total relativistic effects. Spin-orbit coupling contributes at most about
. Again the picture change error is very large, making a difference of about 20 % of the total relativistic effects. The small influence of spin-orbit coupling can be understood as there is effectively only one heavier atom, and this heavy atom is a main group element (see Ref. [Citation59]).
Again the LDA functional performs best of all tested density functionals and results obtained are in excellent agreement with experiment for the diagonal ( deviation) and in good agreement for the off-diagonal tensor components (absolute deviations are of the same size, but relative deviations are between
and
). Correlation effects are on the order of
and thus, contrary to the situation in CHBrClF or the NEQM coupling for Cl, less important than relativistic effects, which amount to about 15 %.
3.3. Discussion
We find surprisingly good results for NEQM coupling tensors of halogen nuclei in chiral methane derivatives when compared to experimental values. We emphasise, however, that we purposely did not use the currently most sophisticated computational approaches to determine the equilibrium structures of these molecule but reused structures determined previously [Citation37] on the CCSD(T) level with a comparatively small basis set to obtain consistent and comparable results for the different molecular properties. Moreover, we did not include any vibrational corrections. Although the latter can be expected to widely cancel out as the sign of nuclear and electronic contributions is opposite, there are cases where vibrational corrections can have a considerable influence on the calculated NEQM coupling tensor [Citation60]. However, at the non-relativistic HF level we could show for an improved molecular structure that the values of the tensor are altered at most on the level of and we do not expect vibrational effects to be much larger. Furthermore, these structure effects suggested even better agreement with experiment if the molecular structure of Ref. [Citation58] is employed. We note in passing that agreement with experiment may also be improved by use of a NEQM that is scaled by the difference between experiment and theory obtained from calculation of a series of similar compounds as done e.g. in Ref. [Citation61] for the NEQMs of
Br and
Br for different methods.
With the methods used herein, larger molecules can be calculated easily, and comparison of CHBrClF and the slightly heavier CHClFI suggests that the performance of the present approaches should improve in molecules with even heavier nuclei for which relativistic effects dominate correlation effects. As usual, some caution has to be taken when using DFT for the estimation of molecular properties, and a range of functionals should be applied in order to allow for an estimate of the importance of electron correlation effects. Results on the performance of different flavours of functionals can vary for different compound classes. For example our results for chiral halomethane derivatives deviate from the findings for mercury halides in Ref. [Citation15], where hybrid functionals were found to outperform LDA and GGA functionals. In other systems, in particular those that contain the transition metal atom Cu, the performance of a variety of density functionals and especially LDA may, however, turn out to be not sufficiently reliable on the whole [Citation62,Citation63]. However, our present study indicates the applicability of 2cZORA-LDA for heavy-element containing chiral main group compounds and motivates future studies of NEQM couplings in these systems with our approach.
4. Conclusion and outlook
In the chiral methane derivatives CHBrClF and CHClFI, which serve as prototypical molecular probes for violations of fundamental symmetries, we have studied nuclear electric quadrupole moment couplings at the level of ZORA-GHF and ZORA-GKS with different density functionals. Our results show good agreement between two-component ZORA-LDA calculations of NEQM couplings and reported experimental values, which is in line with previous studies of main group compounds with DFT and approximate two-component methods. Our methods are easily applicable to larger molecules containing heavier elements and we can expect from our analysis an even improved performance for compounds with heavier nuclei. In future, the method used herein may be applied to other related chiral molecules that are spectroscopically relevant with regard to fundamental aspects of chirality, such as chiral polyhalocubanes [Citation64] or polyhaloadamantanes [Citation65]. Furthermore, this benchmark test enables us to provide support for upcoming microwave experiments with chiral heavy-elemental main group molecules to which more sophisticated methods are not directly applicable. One can also envision our methods to be employed within high-accuracy studies based on composite approaches, in which few single-point calculations for NEQM couplings are performed with coupled cluster methods, whereas vibrational corrections are accounted for with the help of increments determined with the present quasi-relativistic ZORA-DFT methods.
Acknowledgments
This paper is dedicated to Jürgen Gauss on the occasion of his 60th birthday. The authors thank Pekka Pyykkö for enlightening discussions about nuclear quadrupole moments and Melanie Schnell, Daniel Obenchain for raising our interest in electric field gradient calculations of chiral molecules with heavy nuclei. The authors gratefully acknowledge discussion with and help from Jürgen Stohner regarding nuclear electric quadrupole coupling constants in CHBrClF. We are much obliged to Oliver Kreuz, Carsten Zülch and Steffen Giesen for their support in testing our methods. Computer time provided by the Center for Scientific Computing (CSC) Frankfurt is gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- G. Neyens, Rep. Prog. Phys. 66 (4), 633–689 (2003).
- P. Pyykkö, Mol. Phys. 116 (10), 1328–1338 (2018).
- P. Pyykkö and M. Seth, Theor. Chem. Acc. 96 (2), 92–104 (1997).
- L. Visscher, T. Enevoldsen, T. Saue, and J. Oddershede, J. Chem. Phys. 109 (22), 9677–9684 (1998).
- M.G. Kozlov and L.N. Labzowsky, J. Phys. B 28, 1933–1961 (1995).
- K. Gaul, S. Marquardt, T. Isaev, and R. Berger, Phys. Rev. A 99, 032509 (2019).
- K. Gaul and R. Berger, J. Chem. Phys. 152 (4), 044101 (2020).
- S. Stopkowicz and J. Gauss, J. Chem. Phys. 129 (16), 164119 (2008).
- S. Stopkowicz and J. Gauss, J. Chem. Phys. 134 (20), 204106 (2011).
- L. Cheng and J. Gauss, J. Chem. Phys. 134 (24), 244112 (2011).
- M. Pernpointner, M. Seth, and P. Schwerdtfeger, J. Chem. Phys. 108 (16), 6722–6738 (1998).
- J.N.P. van Stralen and L. Visscher, J. Chem. Phys. 117 (7), 3103–3108 (2002).
- J.N.P. van Stralen and L. Visscher, Mol. Phys. 101 (13), 2115–2124 (2003).
- R.L.A. Haiduke, A.B.F. da Silva, and L. Visscher, J. Chem. Phys. 125 (6), 064301 (2006).
- V. Arcisauskaite, S. Knecht, S.P.A. Sauer, and L. Hemmingsen, Phys. Chem. Chem. Phys. 14, 2651–2657 (2012).
- E. van Lenthe and E.J. Baerends, J. Chem. Phys. 112, 8279–8292 (2000).
- I. Malkin, O.L. Malkina, and V.G. Malkin, Chem. Phys. Lett. 361 (3), 231–236 (2002).
- F. Neese, A. Wolf, T. Fleig, M. Reiher, and B.A. Hess, J. Chem. Phys. 122 (20), 204107 (2005).
- F. Aquino, N. Govind, and J. Autschbach, J. Chem. Theory Comput. 6 (9), 2669–2686 (2010). PMID: 26616068.
- J. Autschbach, D. Peng, and M. Reiher, J. Chem. Theory Comput. 8 (11), 4239–4248 (2012). PMID: 26605587.
- V. Arcisauskaite, S. Knecht, S.P.A. Sauer, and L. Hemmingsen, Phys. Chem. Chem. Phys. 14, 16070–16079 (2012).
- C. Puzzarini, G. Cazzoli, A. Baldacci, A. Baldan, C. Michauk, and J. Gauss, J. Chem. Phys. 127 (16), 164302 (2007).
- G. Cazzoli, C. Puzzarini, S. Stopkowicz, and J. Gauss, Mol. Phys. 106 (9–10), 1181–1192 (2008).
- C. Puzzarini, G. Cazzoli, J.C. López, J.L. Alonso, A. Baldacci, A. Baldan, S. Stopkowicz, L. Cheng, and J. Gauss, J. Chem. Phys. 134 (17), 174312 (2011).
- C. Puzzarini, G. Cazzoli, J.C. López, J.L. Alonso, A. Baldacci, A. Baldan, S. Stopkowicz, L. Cheng, and J. Gauss, J. Chem. Phys. 137 (2), 024310 (2012).
- A. Bauder, A. Beil, D. Luckhaus, F. Müller, and M. Quack, J. Chem. Phys. 106, 7558–7570 (1997).
- P. Soulard, P. Asselin, A. Cuisset, J.R.A. Moreno, T.R. Huet, D. Petitprez, J. Demaison, T.B. Freedman, X.L. Cao, L.A. Nafie, and J. Crassous, Phys. Chem. Chem. Phys. 8, 79–92 (2006).
- C. van Wüllen, J. Chem. Phys. 109, 392–399 (1998).
- R. Berger and J. Stohner, Wiley Interdisciplinary Reviews: Computational Molecular Science 9 (3), e1396 (2019).
- V.S. Letokhov, Phys. Lett. A 53, 275–276 (1975).
- O.N. Kompanets, A.R. Kukudzhanov, V.S. Letokhov, and L.L. Gervits, Opt. Commun. 19, 414–416 (1976).
- C. Daussy, T. Marrel, A. Amy-Klein, C.T. Nguyen, C.J. Bordé, and C. Chardonnet, Phys. Rev. Lett. 83, 1554–1557 (1999).
- J.K. Laerdahl, P. Schwerdtfeger and H.M. Quiney, Phys. Rev. Lett 84, 3811–3814 (2000).
- M. Quack and J. Stohner, Phys. Rev. Lett. 84, 3807–3810 (2000).
- A. Soncini, A. Ligabue, P. Lazzeretti, and R. Zanasi, Phys. Rev. E 62, 8395–8399 (2000).
- M. Ziskind, C. Daussy, T. Marrel, and C. Chardonnet, Eur. Phys. J. D 20, 219–225 (2002).
- R. Berger and J.L. Stuber, Mol. Phys. 105, 41–49 (2007).
- J. Cuisset, J.R.A. Moreno, T.R. Huet, D. Petitprez, J. Demaison, and J. Crassous, J. Phys. Chem. A 109, 5708–5716 (2005).
- M. Pernpointner, P. Schwerdtfeger, and B.A. Hess, Int. J. of Quantum Chem. 76 (3), 371–384 (2000).
- P. Fowler, P. Lazzeretti, E. Steiner, and R. Zanasi, Chem. Phys. 133 (2), 221–235 (1989).
- C. van Wüllen, Z. Phys. Chem 224, 413–426 (2010).
- R. Berger, N. Langermann and C. van Wüllen, Phys. Rev. A 71, 042105 (2005).
- R. Berger and C. van Wüllen, J. Chem. Phys. 122, 134316 (2005).
- S. Nahrwold and R. Berger, J. Chem. Phys. 130 (21), 214101 (2009).
- T.A. Isaev and R. Berger, Phys. Rev. A 86, 062515 (2012).
- K. Gaul and R. Berger, J. Chem. Phys. 147 (1), 014109 (2017).
- R. Ahlrichs, M. Bär, M. Häser, H. Horn, and C. Kölmel, Chem. Phys. Lett. 162, 165–169 (1989).
- W. Kohn and L.J. Sham, Phys. Rev. 140, 1133–1138 (1965).
- S.H. Vosko, L. Wilk, and M. Nuisar, Can. J. Phys. 58, 1200–1211 (1980).
- P.A.M. Dirac, Proc. Cambridge Phil. Soc. 26, 376–385 (1930).
- C. Lee, W. Yang, and R.G. Parr, Phys. Rev. B 37, 785–789 (1988).
- A.D. Becke, Phys. Rev. A 38, 3098–3100 (1988).
- P.J. Stephens, F.J. Devlin, C.F. Chabalowski, and M.J. Frisch, J. Phys. Chem. 98, 11623–11627 (1994).
- A.D. Becke, J. Chem. Phys. 98, 1372–1377 (1993).
- A.D. Becke, J. Chem. Phys. 98, 5648–5652 (1993).
- W. Liu, C. van Wüllen, F. Wang, and L. Li, J. Chem. Phys. 116, 3626–3634 (2002).
- L. Visscher and K.G. Dyall, At. Data Nucl. Data Tables 67, 207–224 (1997).
- F. Hobi, R. Berger and J. Stohner, Mol. Phys. 111 (14–15, SI), 2345–2362 (2013).
- R.A. Hegstrom, D.W. Rein, and P.G.H. Sandars, J. Chem. Phys. 73, 2329–2341 (1980).
- M. Seth, M. Pernpointner, G.A. Bowmaker, and P. Schwerdtfeger, Mol. Phys. 96 (12), 1767–1780 (1999).
- W. Bailey and F. Gonzalez, J. Mol. Struct. 651-653, 689–695 (2003).
- P. Schwerdtfeger, M. Pernpointner, and J.K. Laerdahl, J. Chem. Phys. 111 (8), 3357–3364 (1999).
- R. Bast and P. Schwerdtfeger, J. Chem. Phys. 119 (12), 5988–5994 (2003).
- A.A. Fokin, P.R. Schreiner, R. Berger, G.H. Robinson, P. Wei, and C.F. Campana, J. Am. Chem. Soc. 128, 5332–5333 (2006).
- P.R. Schreiner, A.A. Fokin, O. Lauenstein, Y. Okamoto, T. Wakita, C. Rinderspacher, G.H. Robinson, J.K. Vohs and C.F. Campana, J. Am. Chem. Soc. 124 (45), 13348–13349 (2002). PMID: 12418863.