

Abstract
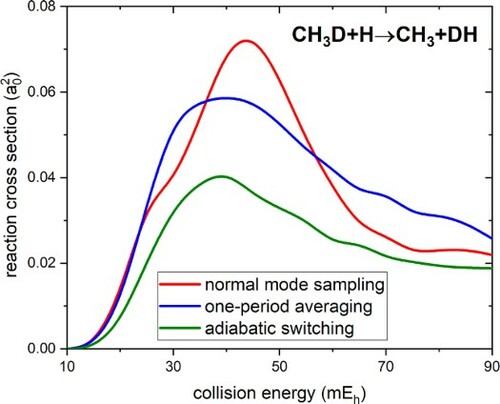
In quasiclassical trajectory simulations of bimolecular reactions of polyatomic molecules, the sets of initial coordinates and momenta of the colliding molecules generated by the widely used normal mode sampling (NMS) are generally nonstationary and evolve during the initial free flight of the reactants. For several isotopologues of the CH4 + H abstraction reaction, the calculated cross sections were found to oscillate as a function of the initial reactant separation. The arising ambiguity can be removed by averaging over an entire period of such oscillations (1PA). Adiabatic switching (AS) produces stationary ensembles of initial states, allowing one to generate unambiguous reaction probabilities and cross sections regardless of the choice of coordinate system used in the zeroth-order harmonic Hamiltonian. In this work the accuracy of the 1PA method was tested against AS-based reactivity parameters. Overall, 1PA provides a reasonably good correction to the non-stationarity of the initial states generated by NMS, but in extreme cases the cross sections are overestimated by up to 70%, thus the accuracy of 1PA varies currently unpredictably and is often sensitive to the choice of coordinates used for NMS. Therefore, it seems advisable to use adiabatic switching to prepare initial states in quasiclassical trajectory simulations.
GRAPHICAL ABSTRACT
Acknowledgements
Financial support to this work has been provided from the National Research, Development and Innovation Fund of Hungary under Grant Nos K129140 (GL), PD120776 and FK134332 (TN). Support by the Hungarian Government, grant number VEKOP2.3.2.-16-2017-00013, co-financed by the European Union is also acknowledged. We acknowledge KIFÜ for awarding us access to computational resources based in Hungary (TN).
Disclosure statement
No potential conflict of interest was reported by the author(s).